
Синдром сэтре чотзена что это

Краткие сведения о нозологии
Выделяют следующие синдромы: синдром Апера, синдром Крузона, синдром Пфайффера, синдром Сэтре–Чотзена, синдром Джексона–Вейса, синдром Карпентера и др.
Клинические синдромы:
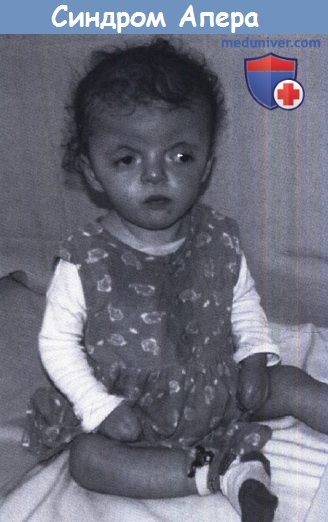
Синдром Апера (Apert syndrome)
Заболевание характеризуется КС коронарных швов с формированием брахицефалии или акроцефалии, вдавленной деформацией средней зоны лица и симметричной синдактилией конечностей. Частота возникновения СА составляет от 7,6 до 22 случаев на 1 млн живорожденных, причем у азиатов частота встречаемости самая высокая, а у испанцев – самая низкая. Наследование СА происходит по аутосомно- доминантному типу и вызывается мутацией гена рецептора фактора роста фибробластов 2-го типа (fibroblast growth factor receptor 2 – FGFR2), расположенного на длинном плече хромосомы 10. Одной из отличительных особенностей пациентов с СА является макрокрания, которая сочетается с КС.
Другой особенностью СА является преждевременное смыкание сфеноокципитального и петроокципитальных синхондрозов.
Нередко у пациентов обнаруживают пороки развития головного мозга, такие как дистопия миндалин мозжечка, стеноз яремного отверстия, арахноидальные кисты в задней черепной ямке, мальформации мозолистого тела и/или лимбических структур.
Следует заметить, что прогрессирующая гидроцефалия неспецифична для заболевания, за нее ошибочно принимают непрогрессирующую вентрикуломегалию, часто наблюдаемую у этих больных. У пациентов с СА обычно наблюдается умственное недоразвитие разной степени выраженности, однако имеются пациенты и с нормальным интеллектом. К основным лицевым признакам СА относится окулярный проптоз (экзорбитизм), который может быть асимметричным. Поражения органа зрения. Гипоплазия верхней челюсти,расщелина язычка или мягкого нёба, расщелина твердого нёба, альвеолярного отростка верхней челюсти и губы очень редки. Задержка прорезывания зубов. Кроме описанных пороков развития, примерно у 2/3 пациентов наблюдается срастание шейных позвонков. Также описаны пороки развития трахеи.
Синдром Крузона (Crouzon syndrome)
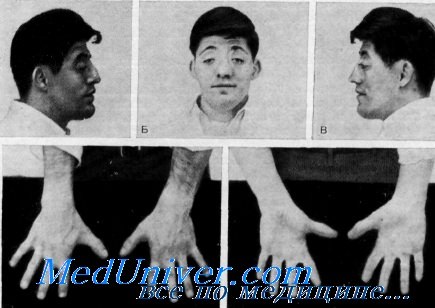
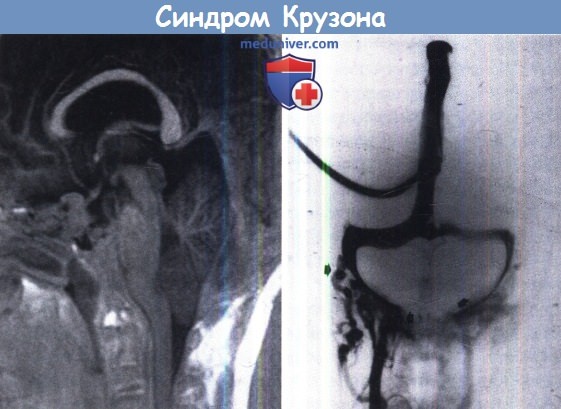
Синдром Крузона (СК) является типичным КС с вовлечением не только коронарного, но и сагиттального и лямбдовидного швов. Порок сопровождается выраженной гипоплазией средней трети лица, с очевидным окулярным проптозом, но при этом заболевании не выявляют грубых пороков развития кистей и стоп, что отличает его отдругих заболеваний этой группы. СК является самым частым в группе синдромальных КС и наблюдается у 1 из 65 000 новорожденных. Так же как и СА он наследуется по аутосомно-доминантному типу и вызывается мутацией гена FGFR2. Обычно к моменту рождения уже синостозировано несколько швов черепа, а в течение первых лет жизни количество включенных в процесс швов может увеличиться. Преждевременное смыкание сфеноокципитального и петрозоокципитального синхондрозов при СК наблюдается часто и происходит в конце внутриутробного периода или вскоре после рождения. Форма головы зависит от того, какие швы и в какой последовательности синостозировались. Кости черепа обычно тонкие, с отчетливыми пальцевыми вдавлениями. Передняя, средняя и задняя черепные ямки короткие, но при этом в отличие от СА практически всегда симметричные. Пороки развития ЦНС наблюдаются у пациентов с СК реже, чем при СА. Они включают аномалию Киари 1-го типа и прогрессирующую гидроцефалию. Стеноз яремных отверстий в сочетании с сужением яремных вен наблюдается в 60% случаев. Более выраженный симметричный экзорбитизм. Верхняя челюсть сильно недоразвита, что в сочетании с костным дефицитом нижнего края орбит усиливает окулярный проптоз. Нёбо узкое, высокое, у половины больных отмечаются латеральные утолщения слизистой оболочки. Нарушения окклюзии и скученность зубов на фоне гипоплазии верхней челюсти так же являются характерными признаками порока, однако в отличие от СА значительной задержки прорезывания зубов нет. Сращение шейных позвонков у 22% больных. Примерно у половины — кондуктивная тугоухость. У 13% имеется стеноз или атрезия наружного слухового канала. Часто наблюдаются значительные нарушения дыхания.
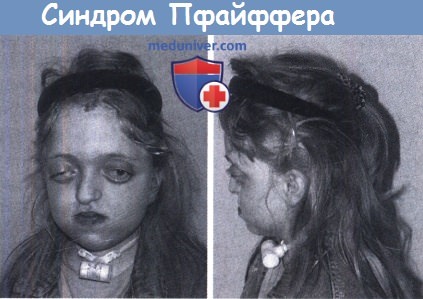
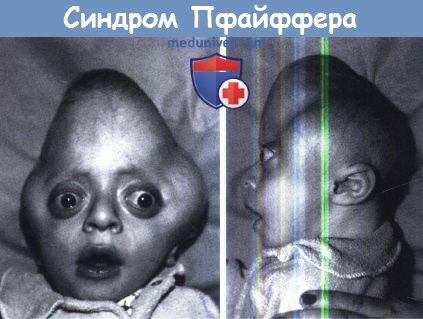
Синдром Пфайффера (Pfeiffer syndrome)
Типичными проявлениями синдрома Пфайффера (СП) являются коронарный КС, гипоплазия
верхней челюсти, экзорбитизм и пороки развития конечностей, такие как широкие большие пальцы
рук и ног, синдактилия и брахидактилия. При СП выявляются мутации генов FGFR1
или FGFR2. Наследуется синдром по аутосомнодоминантному типу. Частота синдрома не
определена из-за его редкости. Выделяют три типа синдрома.
Тип I описывается как классическая или мягкая форма заболевания. Он характеризуется КС коронарного и нередко сагиттального шва, с формированием брахицефалии. Деформация черепа и лица при этом типе напоминает таковую при мягком фенотипе СА. Пациенты с этим типом обычно имеют нормальный или близкий к норме уровень интеллекта и обычную продолжительность жизни.
Тип II более тяжелый и характеризуется КС многих швов. В основном зарастают коронарный, лямбдовидный и метопический швы с компенсаторным ростом черепа по линии открытых сагиттального и височно-теменных швов, что придает голове форму трилистника (череп в виде листа клевера – Cloverleaf skull). При этом типе может отмечаться тяжелый экзорбитизм. Интеллект у детей часто снижен, одним из характерных признаков является плечелучевой синостоз. При отсутствии лечения продолжительность жизни детей с этим типом СП невелика.
Тип III по тяжести черепно-лицевых проявлений похож на тип II, за исключением того, что КС не приводит к формированию трехдольчатого черепа. Большинство пациентов фенотипически напоминают СК, от которого их отличают характерные деформации больших пальцев руки ног. Также у них наблюдается плечелучевой синостоз. Характерными для всех типов СП являются типичные деформации больших пальцев рук и ног в виде их укорочения, утолщения и клинодактилии. Другими признаками являются кожная синдактилия, чаще пальцев стоп и брахидактилия кистей и стоп за счет укорочения средних фаланг. Все пациенты с СП имеют гипоплазию средней зоны лица в сочетании со скученностью зубов и нарушениями прикуса.
Синдром Джексона–Вейса (Jackson–Weiss syndrome)
Синдром Джексона–Вейса характеризуется вовлечением в патологический процесс нескольких швов черепа с формированием акроцефалии. Гипоплазия средней трети лица также типична для этого заболевания, но выражена не так сильно, как при СК. Еще одним характерным признаком являются утолщение и девиация больших пальцев стоп, как при СП, большие пальцы рук при этом не поражаются. Наследуется заболевание по аутосомно-доминантному типу с вариабельной экспрессивностью. Мутация происходит в гене FGFR2. Интеллектуальное развитие часто не страдает. Хотя и описаны индивидуумы с низким или пограничным уровнем интеллекта.
Синдром Карпентера (Carpenter syndrome)
Типичными признаками синдрома Карпентера являются КС и полисиндактилия стоп. В отличие от СА, при котором также описана постаксиальная полидактилия (полидактилия мизинца), при синдроме Карпентера полидактилия всегда преаксиальная. Череп при этом заболевании деформирован по типу акроцефалии и не имеет характерных отличий от других синдромальных КС. Заболевание наследуется по аутосомно-рецессивному типу и его связывают с дефектом RAB23 из группы Ras-генов, который является негативным регулятором сигнальной системы группы НН (hedgehog). Другими признаками болезни являются низкий рост, ожирение, врожденные пороки сердца, задержка интеллектуального развития. Выбор тактики и сроков лечения напрямую зависит от тяжести внутричерепной гипертензии.
Синдром Сэтре–Чотзена (Saethre–Chotzen syndrome)
Синдром Сэтре–Чотзена (ССЧ) единственный из описанных выше синостозов характеризуется асимметрией черепа и лица. Основные проявления ССЧ включают КС, птоз верхних век, высокий лоб с низкой линией роста волос и пороки развития конечностей, такие как брахидактилия и мягкотканная синдактилия. ССЧ наследуется по аутосомно-доминантному типу с высокой пенетрантностью и обусловлен главным образом мутацией гена TWIST. КС наблюдаются у большинства, но не у всех пациентов, чаще всего срастается коронарный шов, приводя к брахицефалии или акроцефалии. Также отмечено зарастание лямбдовидного и метопического швов. Для ССЧ характерны позднее закрытие родничков, увеличенные отверстия теменных выпускников и другие дефекты костей черепа. Длина передней черепной ямки укорочена, а турецкое седло может быть углублено. Из лицевых признаков ССЧ отмечают широкое уплощенное основание носа с искривлением носовой перегородки, нос может быть длинным, тонким, с заостренным или крючковидным кончиком. Верхняя челюсть у таких больных часто недоразвита, что вместе с высоким лбом формирует уплощенный лицевой профиль. Нёбо часто сужено, с высоким сводом и иногда с расщелиной. Аномалии зубов включают сверхкомплектные зубы, гипоплазию эмали и нарушения образования дентина, приводящие к истончению и сужению корней. Деформации конечностей включают частичную мягкотканную синдактилию между II и III пальцами кисти или стопы. Также наблюдаются брахидактилия, клинодактилия и раздвоение дистальных фаланг.
Диагностика:
МСКТ костей черепа и головного мозга с трехмерной реконструкцией наиболее информативный метод диагностики. Позволяет достоверно выявить преждевременно заросший шов, подтвердить, клинически выявленную, деформацию формы головы.
УЗИ костей черепа так же позволяет выявить преждевременное заращение швов черепа.
Показания к оперативному лечению:
- Краниосиностоз
- Повышение внутричерепного давления
- Деформация челюстно-лицевой системы
Противопоказания к оперативному лечению:
- Воспалительный процесс любой локализации в стадии обострения, неполной ремиссии
- Анемия средней и тяжелой степени.
- Тяжелое общесоматическое состояние
- Обострение хронических заболеваний.
Окончательное решение о наличии или отсутствии показаний к оперативному лечению принимает врач-нейрохирург в рамках очной консультации.
Примерное количество койко-дней: 8-10 койко-дней.
Описание результата операции:
Особенности послеоперационного периода:
Ограничения в послеоперационном периоде/
Необходимость в повторной явке в ФЦН. В послеоперационном периоде повторная консультация нейрохирурга не реже 1 раз в 6 мес.
Источник
Глазо-зубо-костная дисплазия. Синдром Пфейффера и Сетре—ЧотценаГлазо-зубо-костная дисплазия — синдром, включающий узкий нос с гипопластичными крыльями, микрокорнеа с аномалией радужной оболочки, синдактилию мягких тканей IV и V пальцев, недостаточность формирования длинных костей и гипоплазию эмали. Среди 45 случаев было 3 больных с проводящей глухотой различной степени (Gorlin et al., Gillespie, Reisner et al.). Синдром Сетре—Чотцена (Saethre—Chotzen) и проводящая глухота. Синдром Сетре — Чотцена характеризуется краииосиностозом, низкой линией роста волос на лбу, асимметрией лица, птозом одного или обоих век, смещением носовой перегородки и разными кожными синдактилиями. Согласно данным Pantkc с соавт., к настоящему времени опубликовано около 85 случаев синдрома. Авторы лично обследовали 31 больного. Bartsocas с соавт. представили хороший литературный обзор.
Pantke сообщил, что у 15% больных имеется легкая проводящая глухота, в некоторых случаях односторонняя. Далее он отметил, что дефект слуха был выявлен у 11 из 22 больных, которым было произведено аудиометрическое обследование. В большинстве этих случаев тип и/или степень глухоты не указаны. Однако Chotzen и Hammar с Roggenkamp отметили проводящую глухоту умеренной степени. Grebe обнаружил глухонемоту, но тип потери слуха не определил. Dolivo и Gillieron сообщили о смешанной глухоте. В наблюдении Jorgenson диагноз является менее определенным, но у ребенка наблюдалась проводящая глухота. Синдром фокальной гипоплазии кожи и смешанной глухоты. Фокальная гипоплазия кожи характеризуется атрофией и линейной гиперпигмептацией поверхностного слоя кожи, ограниченным отложением в коже жира, множественными папилломами слизистых оболочек или кожи вокруг рта, атрофией ногтей и многочисленными скелетными аномалиями. Это заболевание описано Goltz с сотр. и Ginsburg с соавт.. Наследование, вероятно, Х-сцепленное доминантное, летальное для мальчиков. По-видимому, от 5 до 10% больных с этим синдромом имеют дефект слуха. Исследования височной кости не проводились. Holden и Akers сообщили о смешанной глухоте, выявленной у их больного в возрасте 3 лет. St oilman кратко отметил наличие нейросенсорной глухоты у своего больного. Goltz с соавт. также обнаружили смешанную глухоту. Daly, Ginsburg с соавт. и Ferrara описали сужение наружного слухового прохода. Глазо-глоточная мышечная дистрофия и нейросенсорная глухота. Это одно из нескольких заболеваний, при которых наблюдается миопатия лица. Выявляется оно поздно, обычно в середине жизни. Слабость лицевой мускулатуры достигает степени вялого паралича. Вовлечение в процесс жевательных мышц ведет к западению височных областей и отвисанию нижней челюсти. Резко выражен птоз. Лоб обычно испещрен бороздами. Нарастают затруднения в приеме пищи и жидкости. Заболевание характеризуется аутосомyо-доминантным наследованием. В одной семье обнаружена медленно прогрессирующая нейросенсорная глухота (Graf). Conversely, Roberts и Bamforth не отметили глухоты среди 26 больных с этим заболеванием. Kuhn и Еу обнаружили глухоту у 8 из 23 больных с миотонической дистрофией, у 4 из них нарушения слуха возникли в связи с инфекцией среднего уха. – Также рекомендуем “Синдром Варденбурга: клиника, диагностика” Оглавление темы “Наследственные болезни с глухотой”:
|
Источник
Синдромы с краниосиностозом – методы диагностики, лечения по Европейским рекомендациямСиндромные краниосиностозы представляют собой гетерогенную группу патологических состояний, характеризующихся связью раннего слияния нескольких черепных швов и различных врожденных пороков развития, особенно лица и конечностей. Отличительной чертой синдромных черепно-лицевых дизостозов является вовлечение как мозгового черепа (свода и основания), так и лицевого черепа (орбитальная и средняя зоны лицевого скелета). Свод черепа характеризуется множественными синостозами швов и гипоплазией верхней челюсти. До сих пор неизвестно, следует ли часто ассоциированные аномалии развития головного мозга и динамики ликвора, выявляемые при многих из этих состояний, рассматривать как первичные или вторичные проявления. Вероятно, количество и время слияния швов могут играть прямую роль в фенотипе, по крайней мере, при некоторых из этих патологических состояний, о чем свидетельствует более часто встречающееся грыжевое выпячивание миндалин мозжечка при синдроме Крузона чем при синдроме Аперта, последний из которых характеризуется поздним слиянием ламбдовидного шва по сравнению с первым. При этих пороках часто нарушен венозный отток из полости черепа, с развитием вторичной гипоплазии задней черепной ямки и различной степени повышения давления ликвора с расширением субарахноидального или желудочкового пространства. Существует несколько гипотез для объяснения этих изменений: венозная обструкция может быть вторичной по отношению к аномалии роста костей, расстройство может быть первично вызвано ростом диспластического основания черепа или в связи с нарушением венозного оттока у плода и нарушением нормального созревания дренажа задней ямки. Кроме того, при некоторых этих состояниях, в связи с недостаточным развитием средней трети лица появляется экзофтальм с риском повреждения роговицы даже при небольшой травме. В дальнейшем недоразвитие дыхательных путей, в ряде случав, может вызывать изменения функции внешнего дыхания, из которых самыми тяжелыми являются ночные апноэ. Многообразное участие различных структур и черепно-лицевых функций оправдывает общий термин «фациокраниостенозы», указывающий на конкретные ассоциации костных аномалий черепа и лицевого скелета и подчеркивает трудности в их лечении. а) Генетика краниосиностозов. Были выявлены некоторые мутации, ответственные за синдромные краниосиностозы. Обычно они включают три из четырех факторов роста, преимущественно с аутосомно-доминантным типом наследования. При синдромных синостозах один генный дефект порождает различные клинические фенотипы. Предполагается разница во взаимодействии с компонентами внеклеточной матрицы, что могло бы объяснить разнообразия фенотипов при общих дефектах гена.
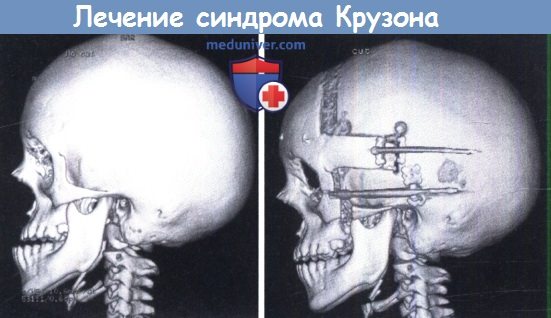
б) Основные синдромы. Краткий обзор основных синдромных краниосиностозов приведен в таблице ниже. 1. Краниосиностоз при синдроме Крузона (черепно-лицевой дизостоз). Синдром Крузона — аутосомно-доминантный синдром, впервые описанный Крузоном в 1912 г. На него приходится около 4,8% случаев краниосиностозов при рождении. Распространенность была оценена как 1:25000 родов. Синдром проявляется краниосиностозом и гипоплазией лица. Фенотип краниосиностоза индивидуально отличен, но в большинстве случаев участвуют коронарные швы. Фенотип лица характерен гипертелоризмом, короткой верхней губой и относительным прогнатизмом нижней челюсти с обратным прикусом. Экзофтальм связан с ретрузией лобной области и верхних челюстей. Эти особенности можно увидеть при рождении, но обычно они появляются в возрасте двух лет с постепенным ухудшением. Тем не менее, существуют некоторые врожденные формы, при которых гипоплазия верхней челюсти является более заметной. Больные страдают от затруднения дыхания и сильного экзофтальма, что может привести к ухудшению смыкания век. Вентрикуломегалия встречается практически всегда, иногда прогрессирующая. Мальформация Киари 1 при этом синдроме, встречается примерно в 70% случаев, может ассоциироваться с сирингомиелией и осложнять хирургическое лечение. Этот порок может быть связан с небольшим размером задней черепной ямки и преждевременным слиянием ламбдовидного шва во время первых двух лет жизни. Ген, ответственный за синдром Крузона расположен на длинном плече хромосомы 10. Более 30 мутаций FGFR2 гена (экзон IIIа и IIIс) были идентифицированы, и они выявляются приблизительно у 60% пациентов. Мутация в FGFR2 также может привести к синдрому Джексона-Вейсса, во многом сходному с синдромом Крузона. Тем не менее, пострадавшие субъекты также предъявляют увеличенные большие пальцы ног и тарзометаnарзальное сращение. Мутации в FGFR3 могут привести к определенной форме синдрома Крузона, связанной с кожными аномалиями (акантокератодермия). 2. Краниосиностоз при синдроме Пфайффера (акроцефалосиндактилия тип V). Синдром был описан в 1964 г. Пфайффером. Частота синдрома Пфайффера была оценена в 1:200000. Он наследуется аутосомно-доминант-ным путем с полной пенетрантностью и варьирующей экспрессией. Синдром Пфайффера — брахицефалия, мембранная синдактилия рук и ног с увеличенными и наклоненными большими пальцами рук и ног. Могут выявляться брахидактилия, анкилоз локтевых суставов и различные висцеральные пороки развития. В настоящее время синдром делится на три подтипа. Подтип 1 является классической и самой легкой формой, с бикоронарным синхондрозом, ведущим к брахицефалии и плоскому лицу, гипертелоризмом и слабой синдактилией с широкими большими пальцами. Подтипы 2 и 3 являются спорадическими и более тяжелыми, с выраженным экзофтальмом и аномалиями мозга, часто, но не всегда связаны с высокой летальностью. Череп в форме листа клевера характеризуется, хотя и не всегда, подтипом 2 с гидроцефалией и маленькой задней черепной ямкой и аномалией Киари 1 типа. Череп в форме листа клевера — признак очень плохого прогноза. Синдром Пфайффера является генетически гетерогенным. Он может быть связан с мутацией в гене FGFR1 или возникать в связи с несколькими типами мутаций в FGFR2. Мутации одного и того же гена, FGFR2, таким образом, могут привести к трем типам фациокраниосиностозов, т. е. Крузона, Джексона-Вейсса и Пфайфера давая основания для предположения об участии в патогенезе и других факторов. 3. Краниосиностоз при синдроме Апера (акроцефалосиндактилии тип I). Синдром Апера является врожденным синдромом, описанным в 1906 г., связывающим фациокраниосиностоз с синдактилией на конечностях. Заболеваемость составляет примерно 1:50000 живорожденных. В большинстве случаев является спорадическим из-за отцовской de novo мутации в экзоне IIIа FGFR2 гена. Описано и аутосомно-доминантное наследование. Краниостеноз является бикоронарным и затрагивает продольной шов. Лицо большое, с гипертелоризмом и экзорбитизмом, нос по типу клюва попугая, прикус перевернутый. Синдактилия может быть полной или с сохранением большого пальца и/или мизинца. Также часто наблюдается подвывих шейных позвонков. Церебральные пороки, в основном с участием мозолистого тела и лимбической структуры, могут быть выявлены с помощью МРТ. На аутопсии было найдено ненормальное развитие височной доли с дисгенезией парагиппокампальной области. Размер желудочков часто увеличен, вентрикуломегалия практически никогда не бывает прогрессирующей. Различная степень умственной отсталости связана с синдромом Апера, около одной пятой пациентов с коэффициентом интеллекта ниже 50, однако также сообщается и о людях с нормальным интеллектом. Высокий уровень психических нарушений связан с аномалиями мозга, в том числе обонятельных-лимбико-септо-каллозальных структур, гипоплазией белого вещества, аномалиями пирамидных путей и непрогрессирующей «ассиметричной» вентрикуломегалией. 4. Краниосиностоз при синдроме Сетре-Хотцена (акроцефалосиндактилии тип III). Первые случаи были зарегистрированы Saethre в 1931 г. и Chotzen в 1932 г.. Более 40% случаев являются семейными. Передача аутосомно-доминантная с неполной пенетрантностью и варьирующей экспрессией. Наиболее типичным фенотипом является брахицефалия и недоразвитие верхней челюсти. Преждевременное слияние черепных швов обычно включает в себя коронарные швы. Тем не менее, может быть вовлечен любой шов с асимметрией лица, необычной формой уха, с частичной синдактилией пальцев рук и ног (второй и третий пальцы, третий и четвертый пальцы с небольшими дистальными фалангами). Большой палец, как правило, увеличен, но не искривлен. Птоз, симметричный или нет, наблюдается почти во всех случаях. Этот синдром может быть чрезвычайно разнообразным в своих клинических проявлениях, таким образом, обследование членов семьи имеет первостепенное значение для выявления потенциальных носителей. Геном, ответственным за синдром Сетре-Хотцена, является TWIST, расположенный на хромосоме 7. Умственная отсталость является редкостью. 5. Краниосиностоз при синдроме Карпентера (акроцефалосиндактилии тип II). Этот синдром, аутосомно-рецессивный, крайне редкий и характеризуется акроцефалией, синдактилией мягких тканей рук, синдактилией и полидактилией ног. Некоторые авторы описывают его с ожирением и гипогонадизмом. 6. Краниосиностоз при синдроме Лежена-Мюнке. Синдром Лежена-Мюнке (Lajeunie-Muenke) характеризуется одно- или двусторонним коронарным синостозом, недоразвитием верхней челюсти, гипертелоризмом и птозом. Передача аутосомно-доминантная. У некоторых пациентов, синдром связан со скелетными аномалиями такими, как наперсткообразные фаланги среднего пальца, конические эпифизы и/или неврологические нарушения, а именно нейросенсорная потеря слуха или умственная отсталость. Несмотря на переменный фенотип, этот синдром был связан с уникальной мутацией в гене FGFR3, Pro 250-Arg. в) Функциональные аспекты. Слияние нескольких швов, которое встречается при синдромном синостозе, приводит к худшему прогнозу интеллектуального развития, усугублению состояния и повышению внутричерепного давления, чем при моношовных синостозах. Более того, аномалии морфологии лица и лицевого роста также могут привести к недостаточной защите глаз, препятствиям дыханию, неправильному прикусу и скученности зубов, которые часто требуют конкретных процедур. Повышение внутричерепного давления (ВЧД) является, по сути, частым для синдромного краниосиностоза. Повышенное ВЧД не только вторично по отношению к синостозу, но и встречается также в сопровождении гидроцефалии и венозных аномалий. Нарушения зрения могут следовать за повышением ВЧД, но они также могут быть вторичными по отношению к экзофтальму, типичному при этих синдромах. Стоит отметить, что атрофия зрительного нерва и потеря зрения наблюдается в основном при синдроме Крузона. Частота отставания умственного развития определяется типом синдрома и сопутствующими пороками развития мозга (в основном нарушения развития прозрачной перегородки). Умственная отсталость часто встречается при синдроме Апера, который представляется наиболее серьезным состоянием, и у некоторых пациентов с синдромом Пфайффера, особенно у пациентов с деформацией черепа в виде листа клевера. С другой стороны, умственная отсталость при синдроме Крузона встречается редко. В общих чертах, когнитивные функции лучше после ранней хирургической коррекции и в случае хорошей психосоциальной среды ребенка. г) Принципы лечения синдромных краниосиностозов. Кранио-фациальная хирургия выполняет задачи как функционального, так и эстетического характера. Первый год жизни имеет первостепенное значение для лечения с целью снижения аномально повышенного внутричерепного давления и, в конечном итоге, нормализации потока ликвора. В самом деле, в первый год, мозг растет очень быстро, и результаты интеллектуального развития гораздо лучше у больных, оперированных на ранних сроках. Дополнительные преимущества раннего хирургического вмешательства включают более легкую реконструкцию костей и высокий потенциал их роста, обеспечивающий «физиологическое» заполнение возможных костных дефектов, которые следуют за хирургической коррекцией. Классическое лечение фациокраниосиностоза включает в себя переднюю реконструкцию черепа, как первый шаг, и коррекцию лица в качестве второго шага. Передний доступ позволяет заниматься как проблемой аномального ВЧД, так и надглазничной рецессией, следовательно, защитить глаза и зрительные функции. Лобно-орбитальное вытяжение может быть выполнено с использованием флотирующей техники или горизонтального вытяжения у детей старше 6-месячного возраста. Улучшение состояния лицевого черепа может быть достигнуто путем остеотомии по типу Le Fort III. В отдельных случаях могут выполняться фронтофациальные моноблочные перемещения с одновременной мобилизацией орбиты и лицевых костей. В настоящее время для достижения адекватного вытяжения лица предпочтительно использовать внутренние или внешние дистракторы. Как правило, вытяжение лица должно быть отложено до тех пор, пока окончательно не сформируются зубные ряды и не возникнет стабильная окклюзия. Тем не менее, в отдельных случаях, особенно в случае нарушения дыхания, может быть необходимо раннее лицевое вытяжение. В других случаях перед хирургическим вмешательством должна быть наложена трахеостома. Раннее расширение заднего черепного свода может также быть выполнено как первый шаг для снижения внутричерепного давления, предполагая фронтофациальную коррекцию до старшего возраста ребенка. Это может быть достигнуто, в частности, при затылочном уплощении, когда задняя ямка чрезмерно мала. Процедура обычно приводит к постепенному улучшению венозного оттока из черепа. Раннее заднее расширение может быть достигнуто как увеличением теменно-затылочного костного лоскута, так и применением пружинной краниопластики у детей с открытым лямбдовидным швом. Расширение БЗО также может быть необходимо в некоторых случаях с симптомной мальформацией Киари 1 типа. Расширение желудочков, встречающееся при синдромном краниосиностозе также может потребовать лечения. Часто это связано с грыжей миндаликов различной степени, и расширение БЗО может стать необходимым. В других случаях, с механическим препятствием току ликвора, эндоскопическая вентрикулоцистерностомия может обеспечить необходимый контроль ВЧД. Тем не менее, в ряде случаев синдромного краниосиностоза венозный отток нарушается. Прогрессирующее открытие коллатерального венозного канала может привести к отсутствию гидроцефалии. Тем не менее, некоторым пациентам может потребоваться установка вентрикулоперитонеального шунта. Во всех случаях требуется несколько видов хирургического вмешательства. Тесное сотрудничество между пластическим хирургом и нейрохирургом, а также детским нейроанестезиологом является обязательным условием получения хорошего функционального и косметического исхода.
– Также рекомендуем “Нейрофиброматоз 1 типа – методы диагностики, лечения по Европейским рекомендациям” Оглавление темы “Краниосиностоз.”:
|







Источник