
Синдром свайера и лимфангиолейомиоматоз что это

Синдром Свайера – это нарушение формирования пола, характеризующееся кариотипом 46XY, врождённой дисгенезией гонад при первично сформированных прочих женских гениталиях – влагалище, матке, фаллопиевых трубах. К признакам патологии относятся первичная аменорея, маскулинное телосложение, половой инфантилизм. Диагноз выставляется на основании анамнестических данных, результатов общего и гинекологического осмотра, интроскопических методов исследования органов малого таза, гормонального и молекулярно-генетического анализа. Лечение состоит из двух этапов – удаления неразвитых половых желёз и длительного применения заместительной гормональной терапии.
Общие сведения
Синдром Свайера (полную, или «чистую» дисгенезию гонад) можно кратко охарактеризовать как женский фенотип при мужском генотипе. Заболевание названо по имени британского эндокринолога Джералда Свайера, описавшего его в 1955 году как случай мужского псевдогермафродитизма. Полная форма дисгенезии является несиндромальной (не сопровождается экстрагенитальными пороками развития), исключает двойственность полового развития (наличие мужских первичных половых признаков наряду с женскими), психологическое развитие происходит по женскому типу. Врождённая патология встречается в одном случае на 20 000 индивидов с мужским кариотипом и регистрируется чаще иных форм XY-дисгенезии гонад.
Синдром Свайера
Причины
Этиология синдрома Свайера изучена недостаточно. На сегодняшний день известно, что чаще всего возникновение патологии связано с отсутствием или мутацией гена SRY, расположенного на коротком плече хромосомы Y и большей частью отвечающего за контроль формирования тестикул. Судя по наблюдениям семейных случаев заболевания, возможна причастность пока неизвестных X-сцепленных или аутосомных генов.
Факторы риска тоже до конца не установлены. Кроме общих мутагенных воздействий (ионизирующего излучения и интоксикаций, вирусных инфекций, несбалансированного или пониженного питания) и уже упомянутого наследственного отягощения предполагается, что вероятность патологии может находиться в прямой зависимости от возраста отца. Чаще всего проследить связи между каким-либо воздействием на организм во время гестации и развитием синдрома Свайера не удаётся.
Патогенез
Формирование репродуктивных органов происходит из мюллерова протока у женщин и вольфова – у мужчин. У эмбриона с мужским генотипом синтез мужских стероидных гормонов клетками Лейдига в эмбриональных семенниках обусловлен воздействием материнского хорионического гонадотропина. Клетки Сертоли стимулируют дифференцировку клеток Лейдига и прочих, продуцируют антимюллеров гормон, способствующий атрофии мюллерова протока. Их нормальная деятельность обуславливает развитие индивида мужского пола – с адекватной дифференцировкой тестикул, атрофией мюллерова протока.
Выраженные сбои этого механизма приводят к формированию из бипотентных зачатков женских репродуктивных органов, развитие которых не требует столь сложной регуляции. Созревание мужского эмбриона контролируется геном SRY. При его отсутствии или мутации нарушается деятельность клеток Сертоли, дифференцировка гонад не происходит, что влечёт развитие синдрома Свайера – фенотипически женского организма без полноценных яичников, способных в дальнейшем стимулировать развитие вторичных половых признаков, но с бесполезными, склонными к малигнизации зачатками желёз.
Симптомы
В допубертатном периоде патология протекает без каких-либо субъективных проявлений. В период пубертата для синдрома Свайера характерно отсутствие признаков полового созревания. Может отмечаться лишь скудное оволосение в области лобка и подмышек, однако нередко отсутствует и оно. Менархе не наступает, молочные железы не развиваются или выражены очень слабо. Тип телосложения при овариальной дисгенезии мужской – с широкими плечами, объёмной грудной клеткой, узким тазом.
У женщин с синдромом Свайера чаще наблюдается нормальный или выше среднего рост, развитая мускулатура, «тяжёлая» нижняя челюсть. Иногда возникает незначительная гипертрофия клитора, хотя обычно наружные гениталии несколько недоразвиты. Больные жалуются на первичное бесплодие, ощущение дискомфорта или боли ввиду недостаточного развития вагины при половом акте или гинекологическом осмотре.
Осложнения
Частым (у 20-60% пациенток) осложнением синдрома Свайера является развитие опухолей, по большей части исходящих из рудиментарного полового тяжа, которым, по сути, представлены дисгенетичные гонады – дисгермином, андробластом. Эти неоплазии нередко имеют злокачественный характер, возникают с обеих сторон (синхронно или метахронно), регистрируются в раннем репродуктивном, подростковом и детском возрасте.
К последствиям нелеченного синдрома Свайера также можно отнести раннее появление патологий, обусловленных дефицитом эстрогена у женщин – остеопороза, сердечно-сосудистых заболеваний (артериальной гипертензии, ишемической болезни сердца). Кроме того, для многих больных диагноз становится источником тяжёлого психологического страдания по поводу «утраты женственности», а у замужних женщин – и боязни распада семьи.
Диагностика
Диагностика синдрома Свайера осуществляется гинекологом при участии медицинского генетика. Чаще всего диагноз устанавливается в 14-15 лет при обращении к врачу по поводу отсутствия полового развития, иногда – позднее, по причине бесплодия. В некоторых случаях первым признаком заболевания и основанием для углублённого исследования является опухолевое образование рудиментарных половых желёз, случайно обнаруженное врачом или самой больной. Диагностические мероприятия включают:
- Клинический осмотр. В ходе общего осмотра выявляется отсутствие основных вторичных женских половых признаков на фоне нормального или ускоренного роста. При гинекологическом осмотре обнаруживаются нормально сформированные, но недоразвитые женские наружные половые органы. Косвенным подтверждением диагноза является наличие кровных родственниц с полной или частичной дисгенезией гонад.
- Лучевые методы исследования. Наиболее доступным и информативным методом диагностики является гинекологическое УЗИ. Ультрасонография позволяет обнаружить такие характерные для патологии объективные признаки, как гипоплазию матки, извитые, недоразвитые маточные трубы, тяжевидные «яичники» (иногда – с новообразованиями). В сомнительных случаях назначается МРТ.
- Гормональный анализ. Характерным признаком синдрома Свайера считается значительное повышение гонадотропных (фолликулостимулирующего, лютеинизирующего) гормонов в сыворотке крови, снижение уровня эстрадиола. Гестагеновая функциональная проба отрицательная, циклическая (эстроген-гестагенная) гормональная проба положительная.
- Генетическое тестирование. Применяется с целью верификации диагноза. Цитогенетическое исследование выявляет мужской кариотип (46, XY), а молекулярно-генетическое – отсутствие или повреждение гена SRY. При невозможности генетического обследования назначается гистологическое исследование биоптата яичников. Диагноз подтверждает соединительнотканная структура с отсутствием фолликулов.
Синдром Свайера дифференцируют с другими формами дисгенезии гонад – синдромом Шерешевского-Тёрнера, мозаицизмом 45,X/46,XY, синдромом Морриса (синдромом тестикулярной феминизации), а также с задержкой полового развития гипоталамо-гипофизарного, конституционального, идиопатического генеза. При подозрении на опухоль необходима консультация онкогинеколога, а для выявления центральных форм задержки полового созревания – эндокринолога.
Лечение синдрома Свайера
Консервативная терапия
Лечение заболевания направлено на предупреждение осложнений, нормализацию психоэмоционального статуса пациентки, по возможности – на реализацию репродуктивной функции. После установления диагноза осуществляется хирургическая операция (удаление рудиментарных гонад), затем назначается гормональная терапия. Раннее (с подросткового периода) начало лечения синдрома Свайера повышает вероятность вынашивания ребёнка.
- Заместительная гормональная терапия. Проводится чередованием эстрогенных и прогестиновых препаратов в течение менструальных циклов. Назначается с периода полового созревания до возраста климактерия. Лечение нормализует течение метаболических процессов, способствует развитию матки, женских половых вторичных признаков, гравидарной подготовке эндометрия.
- Коррекция нервно-психических нарушений. Психотерапевтическое воздействие на личность позволяет перестроить отношение больной к себе и ближайшему окружению, преобразовать собственные психологические установки, снять психоэмоциональное напряжение. Иногда лечение дополняется фармакотерапией (антидепрессантами, транквилизаторами).
При достаточном развитии матки возможно применение программы ЭКО с имплантацией донорских ооцитов, оплодотворённых спермой супруга пациентки. На протяжении вынашивания плода женщине назначается сначала поддерживающая, затем сохраняющая гормонотерапия. Родоразрешение осуществляется оперативно. В современных репродуктологии и акушерстве уже накоплен успешный опыт ведения беременности у больных с дисгенезией гонад (в том числе синдромом Свайера).
Хирургическое лечение
Оперативное вмешательство в объёме удаления рудиментарных гонад и маточных труб выполняется непосредственно после диагностики этой формы врождённой овариальной агенезии. Хирургическое лечение проводится с целью предотвращения новообразований, источником которых являются клетки дисгенетичных половых желёз. Консервативное лечение не рекомендуется начинать до хирургической операции, поскольку гормональная терапия повышает риск развития онкологических осложнений.
Прогноз и профилактика
При своевременно начатом рациональном лечении синдрома Свайера прогноз относительно продолжительности и качества жизни благоприятный. Некоторые пациентки могут реализовать детородную функцию с помощью вспомогательных репродуктивных технологий, описаны случаи повторных успешных беременностей. Первичная профилактика рождения девочек с XY-дисгенезией включает здоровый образ жизни во время беременности, исключение профессиональных и бытовых вредностей. Для раннего выявления и лечения заболевания следует подвергать генетическому обследованию монозиготных близнецов больных.
Источник
Синдром Свайера – это довольно редко встречающееся врожденное заболевание, развитие которого свидетельствует о нарушении структуры игрек-хромосомы (речь идет об отсутствии определенного гена или его изолированой мутации).
Причины развития заболевания
Как правило, непосредственной причиной возникновения данного заболевания является точечная мутация определенного гена, находящегося в коротком плече игрек-хромосомы, или полная утрата этого гена. Данный участок хромосомы отвечает за синтез белка, принимающего участие в развитии пола эмбриона по мужскому типу. В результате, поскольку воздействие мужских половых гормонов отсутствует, плоду остается единственный вариант – формироваться по женскому типу. В результате родившийся ребенок имеет женский фенотип при кариотипе “XY”.
Патогенез
Мутации или отсутствие гена SRY ведет к сбою в дифференциации клеток Сертоли, и, как следствие, к недоразвитию семенных канальцев.
В результате, невзирая на «мужской» кариотип XY, половые органы плода закладываются и формируются по женскому типу.
Клинические проявления
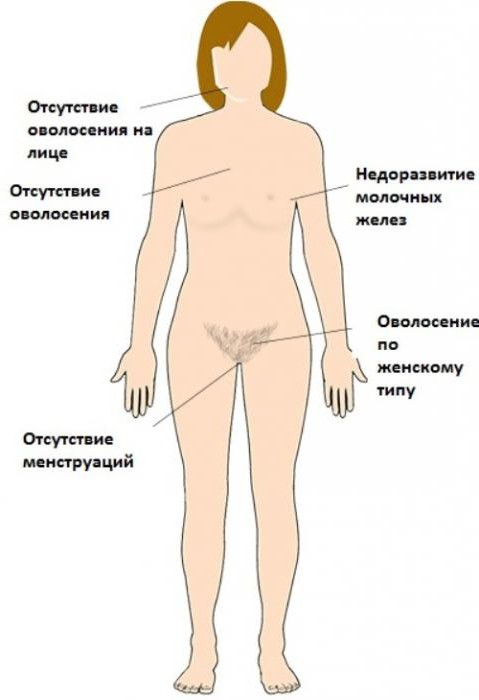
Вплоть до начала пубертатного периода признаки синдрома Свайера внешне практически не выражены. И лишь по мере взросления девочки начинают проявляться определенные особенности:
- Недостаточность оволосения подмышечных впадин и области внешних половых органов.
- Недостаточное, слабое развитие молочных желез.
- Различной степени недоразвитие, инфантилизм матки.
- Гипоплазия влагалища (встречается реже).
- Неярко выраженные вторичные половые признаки – «евнухоподобный» или интерсексуальный тип телосложения.
- Гипотрофия или атрофия слизистой оболочки половых органов.
- Недоразвитие наружных половых органов (половых губ и клитора).
- Генитальный инфантилизм.
- В ряде случаев наблюдается чрезмерно активный рост тела и его отдельных частей: нижней челюсти, плечевого пояса (в результате чего формируются широкие плечи), мышечной массы.
- Наступление полового созревания у девушек с синдромом Свайера невозможно ввиду отсутствия в их организме эстрогенов.
- Полная стерильность.
Диагностика
В подавляющем большинстве случаев заболевание диагностируется в возрасте 15-16 лет, в период полового созревания, когда становится очевидно, что у пациентки не выражены вторичные половые признаки.
В это же время девушки, имеющие такую мутацию, достигнув этого возраста, начинают обращаться к гинекологу с жалобами на задержку наступления менструаций.
Иногда диагностика осуществляется в результате дисплазии и малигнизации недоразвитых гонад.
Постановка диагноза «синдром Свайера» основывается на следующих факторах:
- Физикальное обследование пациентки.
- Ультразвуковое обследование.
- Гистеросальпингография.
Однако подтверждение диагноза возможно лишь при помощи исследования полового хроматина, которое выявляет наличие мужского кариотипа при женском фенотипе.
Возможности лечения
Лечение синдрома Свайера осуществляется в нескольких направлениях.
- В первую очередь производится удаление яичников – ввиду высокой вероятности преобразования их в злокачественные опухоли.
- После овариоэктомии назначается заместительное лечение при помощи гормональных препаратов. Это способствует развитию вторичных половых признаков.
- В случае, когда матка развита достаточно, существует возможность вынашивания и рождения здорового ребенка (беременность наступает в результате экстракорпорального оплодотворения).
Данное заболевание следует отличать от сходного по названию синдрома Свайера-Джеймса-МакЛеода. Это состояние, как и сходный по проявлениям лимфангиолейомиоматоз, является патологией, поражающей легочную ткань. Синдром Свайера и лимфангиолейомиоматоз – разные заболевания.
Источник
Медицинский эксперт статьи
х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Лимфангиолейомиоматоз (лейомиоматоз) представляет собой доброкачественную пролиферацию гладкомышечных клеток во всех отделах легкого, в том числе легочных кровеносных и лимфатических сосудов, а также плевры. Заболевание лимфангиолейомиоматоз (лейомиоматоз) – редкое, встречается исключительно у молодых женщин. Причина неизвестна. Проявляется одышкой, кашлем, болью в грудной клетке и кровохарканьем; часто развивается спонтанный пневмоторакс.
Подозрение на наличие заболевания устанавливается при анализе клинических проявлений и результатов рентгенографии органов грудной клетки и подтверждается при КТ высокого разрешения. Прогноз не установлен, но болезнь медленно прогрессирует, и в течение ряда лет часто приводит к дыхательной недостаточности и смерти. Радикальное лечение лимфангиолейомиоматоза – трансплантация легкого.
Лимфангиолейомиоматоз (лейомиоматоз) диссеминированный – патологический процесс, характеризующийся опухолевидным разрастанием гладкомышечных волокон по ходу мелких бронхов, бронхиол, стенок кровеносных и лимфатических сосудов легких с последующей мелкокистозной трансформацией легочной ткани. Заболевание поражает только женщин в возрасте 18-50 лет.
[1], [2], [3], [4], [5], [6], [7], [8]
Что вызывает лимфангиолейомиоматоз?
Лимфангиолейомиоматоз – болезнь легких, поражающая исключительно женщин, преимущественно в возрасте между 20 и 40 годами. Представительницы белой расы находятся в группе наибольшего риска. Лимфангиолейомиоматоз встречается с частотой менее 1 случая на 1 млн человек и характеризуется доброкачественной пролиферацией атипичных гладкомышечных клеток в грудной клетке, включая паренхиму легкого, кровеносные и лимфатические сосуды и плевру, приводя к изменению структуры легкого, кистозной эмфиземе и прогрессирующему снижению функции легкого. Описание данного заболевания включено в текущую главу, поскольку больным лимфангиолейомиоматозом иногда ошибочно устанавливается диагноз ИБЛАРБ.
Причина лимфангиолейомиоматоза неизвестна. Привлекательная гипотеза о том, что женские половые гормоны играют роль в патогенезе заболевания, остается недоказанной. Лимфангиолейомиоматоз обычно возникает спонтанно и во многом схоже с туберозным склерозом. Лимфангиолейомиоматоз встречается также у некоторых больных ТС; поэтому было высказано предположение о том, что он является особой формой ТС. Мутации комплекса генов туберозного склероза-2 (TSC-2) были описаны в клетках лимфангиолейомиоматоза и в ангиомиолипомах. Эти данные свидетельствуют о наличии одной из двух возможностей:
- Соматическая мозаичность мутаций TSC-2 в легких и почках приводит к появлению очагов заболевания среди нормальных клеток этих тканей (в то время как можно ожидать появление отдельных участков заболевания);
- Лимфангиолейомиоматоз представляет собой распространение ткани ангиомиолипомы в легкое, аналогичное наблюдаемому при синдроме доброкачественной метастазирующей лейомиомы.
Характерны следующие патоморфологические признаки заболевания:
- значительная плотность легких, множество мелких узелков 0.3-0.7 см в диаметре, белесоватых, заполненных жидкостью, они расположены субплеврально;
- наличие в отдельных участках легких крупных воздушных полостей;
- гиперплазия лимфоузлов;
- диффузная пролиферация гладкомышечных волокон в интерстиции легких (межальвеолярно, периваскулярно, перибронхиально, субплеврально, по ходу лимфатических сосудов);
- деструктивные изменения стенок кровеносных и лимфатических сосудов, стенок бронхов, альвеол;
- формирование микрокистозного «сотового» легкого;
- развитие пневмогемохилотрокса в связи с деструкцией стенок кровеносных и лимфатических сосудов легких и разрывом субплевральных кист.
Указанные патоморфологические проявления характерны для диффузной формы заболевания.
Очаговая форма (лейомиоматоз) характеризуется развитием в паренхиме легких опухолевидных образований – лейомиом.
Симптомы лимфангиолейомиоматоза
Начальными проявлениями заболевания являются одышка и, реже, кашель, боли в грудной клетке и кровохарканье. В целом, проявления обычно скудные, но у некоторых пациенток могут отмечаться влажные и сухие хрипы. Часто развивается спонтанный пневмоторакс. Возможны также проявления обструкции лимфатического протока, включая хилоторакс, хилезный асцит и хилурию. Ухудшение, как считается, наступает во время беременности и, возможно, при авиаперелетах; последние особенно противопоказаны при появлении или усугублении проявлений со стороны дыхательной системы; наличии в анамнезе пневмоторакса или кровохарканья и признаков обширных субплевральных буллезных или кистозных изменений, выявленных при КТВР. Ангиомиолипомы почек (гамартомы, состоящие из гладких мышц, кровеносных сосудов и жировой ткани) встречаются у 50 % пациентов и, хотя обычно протекают бессимптомно, могут вызвать кровотечение, которое, при прогрессировании, обычно проявляется в виде гематурии или боли в области фланка.
Длительное время заболевание протекает бессимптомно. В развернутой стадии характерные симптомы лимфангиолейомиоматоза:
- одышка, вначале она беспокоит только при физической нагрузке, в дальнейшем становится постоянной;
- боли в грудной клетке, усиливающиеся при дыхании;
- кровохарканье (непостоянный симптом);
- рецидивирующий спонтанный пневмоторакс – наблюдается у 1/3-1/2 больных, проявляется внезапной интенсивной болью в грудной клетке, одышкой, отсутствием везикулярного дыхания и тимпаническим оттенком перкуторного звука на стороне поражения;
- хилоторакс – скопление хилезной жидкости в плевральной полости (с одной или с обеих сторон). При развитии хилоторакса усиливается одышка, появляется интенсивный тупой звук при перкуссии над областью выпота, дыхание в этом месте отсутствует; хилезная жидкость накапливается снова после ее удаления. Характерно, что развитие пнево- и хилоторакса совпадает с менструацией;
- хилоперикардит и хилезный асцит развиваются по мере прогрессирования заболевания и их появление также совпадает с менструациями;
- развитие хронического легочного сердца (симптоматику см. «Легочная гипертензия»).
Очаговая форма заболевания протекает бессимптомно и выявляется рентгенологически. В некоторых случаях заболевание принимает системный характер – лейомиомы развиваются в брюшной полости, забрюшинном пространстве, матке, кишечнике, почках.
Активизации заболевания способствуют беременность, роды, прием контрацептивов.
Диагностика лимфангиолейомиоматоза
Подозрение на наличие лимфангиолейомиоматоза возникает у молодых женщин, предъявляющих жалобы на одышку, при наличии интерстициальных изменений при нормальном или увеличенном объеме легких по данным рентгенографии органов грудной клетки, спонтанного пневмоторакса, и/или хилезном выпоте. Верификация диагноза возможна при проведении биопсии, но сначала во всех случаях должна выполняться КТВР. Выявление множественных мелких диффузно распределенных кист является патогномоничным для лимфангиолейомиоматоза.
Биопсия выполняется только при наличии сомнений по результатам КТВР. Обнаружение при гистологическом исследовании патологической пролиферации гладкомышечных клеток (клеток лимфангиолейомиоматоза), ассоциированной с кистозными изменениями, является подтверждением наличия заболевания.
Результаты исследований функции легких также свидетельствуют в пользу диагноза и особенно полезны в контроле за динамикой процесса. Типичные изменения заключаются в появлении обструктивного или смешанного (обструктивного и рестриктивного) типа нарушений. Легкие обычно гипервоздушны, с повышением общей емкости легких (ОЕЛ) и воздушности грудной клетки. Обычно отмечается задержка воздуха (увеличение остаточного объема (ОО) и отношения ОО/ОЕЛ). Также обычно снижаются РаО2 и диффузионная способность в отношении монооксида углерода. У большинства пациентов также выявляется снижение работоспособности.
[9], [10], [11], [12]
Лабораторная диагностика лимфангиолейомиоматоза
- Общий анализ крови – существенных изменений нет. У некоторых больных имеется эозинофилия, нередко увеличивается СОЭ, особенно при развитии пневмо-хилоторакса.
- Общий анализ мочи – может наблюдаться незначительная протеинурия (симптом неспецифичный и непостоянный);
- Биохимический анализ крови – иногда наблюдается гиперхолестеринемия, возможно увеличение уровня альфа2- и гамма-глобулинов, аминотрансфераз, общей лактатдегидрогеназы, ангиотензинпревращающего фермента.
- Исследование плевральной жидкости. Хилоторакс чрезвычайно характерен для лимфангиолейомиоматоза. Плевральная жидкость имеет следующие характерные особенности:
- цвет молочно-белый;
- мутность жидкости сохраняется после центрифугирования;
- содержание триглицеридов больше 110 мг%;
- содержит хиломикроны, которые выявляются при электрофорезе липопротеинов в полиакриламидном геле.
Инструментальная диагностика лимфангиолейомиоматоза
- Рентгенологическое исследование легких. При диффузной форме заболевания характерными рентгенологическими признаками являются усиление легочного рисунка за счет развития интерстициального фиброза и диффузные множественные мелкоочаговые (милиарные) затемнения. В последующем в связи с формированием множества мелких кист появляется картина «сотового легкого».
Для очаговой формы характерны очаги затемнения от 0.5 до 1.5 см в диаметре с четкими границами.
При развитии пневмоторакса определяется спавшееся поджатое воздухом легкое, при развитии хилоторакса – интенсивная гомогенная тень (за счет выпота) с косой верхней границей.
- Компьютерная томография легких выявляет те же изменения, но значительно раньше, в том числе кистоподобые и буллезные образования.
- Исследование вентиляционной способности легких. Характерно увеличение остаточного объема легких в связи с образованием множества кист. У большинства больных определяется также обструктивный тип дыхательной недостаточности (снижение ОФВ1). По мере прогрессирования заболевания присоединяется также рестриктивная дыхательная недостаточность (снижение ЖЕЛ).
- Исследование газов крови. По мере развития дыхательной недостаточности появляется артериальная гипоксемия, парциальное напряжение кислорода снижается, особенно после физической нагрузки.
- ЭКГ. По мере прогрессирования заболевания выявляются признаки гипертрофии миокарда правого предсердия и правого желудочка (см. «Легочная гипертензия»).
- Биопсия легкого. Исследование биоптата легочной ткани производится для верификации диагноза. Информативна только открытая биопсия легкого. В биоптате выявляются диффузная пролиферация гладкомышечных волокон в интерстиции легких.
Программа обследования при лимфангиолейомиоматозе
- Общие анализы крови, мочи.
- Биохимический анализ крови – определение содержания холестерина, триглицеридов, общего белка, белковых фракций, билирубина, трансаминаз.
- Рентгенологическое исследование легких.
- Исследование плевральной жидкости – оценка цвета, прозрачности, плотности, цитология, биохимический анализ (определение холестерина, триглицеридов, глюкозы).
- ЭКГ.
- УЗИ органов брюшной полости и почек.
- Открытая биопсия легкого с последующим гистологическим исследованием биоптатов.
Какие анализы необходимы?
Лечение лимфангиолейомиоматоза
Стандартное лечение лимфангиолейомиоматоза – трансплантация легкого, но заболевание может рецидивировать и в трансплантате. Альтернативные методы лечения, в частности гормональная терапия прогестинами, тамоксифеном и овариэктомия, являются в большинстве случаев неэффективными. Пневмоторакс также может представлять проблему, поскольку может часто рецидивировать, быть двусторонним и нечувствительным к стандартной терапии. Повторные пневмотораксы требуют декортикации легкого, плевродеза (тальком или другими веществами) или плеврэктомии. В США пациенты могут получить психологическую поддержку в Фонде лимфангиолейомиоматоза.
Какой прогноз имеет лимфангиолейомиоматоз?
Лимфангиолейомиоматоз имеет не ясный прогноз, ввиду крайней редкости заболевания и значительной вариабельности клинического состояния больных лимфангиолейомиоматозом. В целом, заболевание медленно прогрессирует, приводя, в конечном счете, к дыхательной недостаточности и смерти, но ожидаемая продолжительность жизни широко варьирует по данным различных источников. Пациентки должны знать, что прогрессирование заболевания может ускориться во время беременности. Медиана выживаемости составляет приблизительно 8 лет с момента постановки диагноза.
Источник