
Синдром задних уретральных клапанов у плода

Девочки нужны положительные примеры.
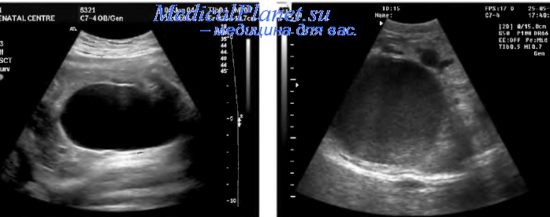
Сестра мужа, Б 17-18 нед. Поставили диагноз мегацистис плода, дилатация уретры 2 степени, синдром задних уретральных клапанов, синдром prune-belly, гиперплазия плаценты, выраженное маловодие.
Есть ли шанс, что все будет хорошо? Или готовиться к прерыванию?
Источник

Клапаны уретры — это врожденная, реже приобретенная патология, при которой просвет мочеиспускательного канала обтурируется складчатыми образованиями слизистой. Проявляется ослаблением струи мочи, умеренной болезненностью при мочеиспускании, энурезом, поллакиурией, увеличением мочевого пузыря. Диагностируется с помощью цистоуретрографии, уретроскопии, уродинамического исследования, бужирования уретры. Лечение хирургическое путем эндоуретральной или промежностной электрорезекции клапанных образований, перед проведением операции возможно наложение разгрузочной цистостомы или уретерокутанеостомы.
Общие сведения
Врожденные клапаны уретры выявляются у 0,002-0,015% мальчиков, более чем в половине случаев диагностируются на первом году жизни. Редко уретральные клапанные структуры определяются у девочек с адреногенитальным синдромом и маскулинизированным мочеиспускательным каналом. В спорадических случаях дефект слизистой является приобретенным и обнаруживается у взрослых. Аномально развитые складки могут быть единичными и множественными. Врожденный вариант дефекта слизистой уретры часто наблюдается в структуре малых врожденных пороков развития, диабетической фетопатии, сочетается с нейрофиброматозом, сирингомиелией, VATERL/VACTERL-синдромом, стенозом мочеточников, контрактурой мочепузырной шейки, гипертрофией семенного бугорка, гипоплазией и эктопией яичек, атрофией простаты и др.

Клапаны уретры
Причины
Формирование аномальных клапанных образований в задних отделах мочеиспускательного канала связано с ненаследственным нарушением ранних этапов эмбриогенеза органов мочевыделительной системы. Точная этиология этого варианта дизэмбриогенеза уретры пока не установлена. По мнению специалистов в сфере современной урологии, вероятными причинами возникновения задних уретральных клапанов являются:
- Генетические мутации. Хотя наследственный характер заболевания не доказан, нормальное развитие мочевыводящих органов может нарушаться при повреждении ДНК материнской и отцовской клеток, образовавших зиготу. Теория генетически детерминированного дизэмбриогенеза подтверждается более частым выявлением дефекта у детей возрастных пар и у родителей, которые злоупотребляют психоактивными веществами или длительно работают на вредных производствах с возможными мутагенными влияниями.
- Повреждающие воздействия в период беременности. Неправильная закладка мочеиспускательного канала чаще выявляется у мальчиков, рожденных женщинами, которые в I триместре перенесли простудное заболевание с высокой температурой. Роль инфекционного патогена в возникновении аномалии связана с задержкой отдельных этапов формирования уретры, нарушением дифференцировки клеток, гиперплазией тканей. Наиболее тератогенными считаются возбудители краснухи, герпеса, цитомегаловирусной инфекции.
С учетом стимулирующего действия тестостерона на формирование уретрального канала у мальчиков не исключено дизэмбриогенетическое влияние приема беременной антиандрогенных гормональных препаратов. Формирование передних врожденных уретральных клапанов становится результатом действия тех же факторов, что и задних. В редких случаях обтурирующие складки слизистой бульбарного и пенального отделов уретры являются приобретенными и образуются при грубом бужировании уретры, нарушении техники уретроскопии, цистоскопии либо являются следствием операций по поводу гипоспадии.
Патогенез
Механизм образования задних клапанов уретры связан с неполной инволюцией урогенитальной мембраны или неправильным внедрением вольфова канала в стенку мочевыводящего отдела. При дизонтогенезе мезонефральный проток проходит не в задней стенке мочеиспускательного канала, а проникает через его переднюю стенку, образуя перегородки и клапаны, нарушающие отток мочи. Гипертрофированные складки, перекрывающие уретру, также возникают при неполной редукции мочеполового участка клоакальной мембраны. Аномалия формируется на 4-6-й неделях гестационного срока при разделении первичной клоаки на ректальный и мочевыделительный отделы.
На 11-й неделе онтогенеза после начала выделения мочи почками плода развивается инфравезикальная обструкция мочевыделения разной степени выраженности. При значительном перекрытии уретры клапанами мочеиспускательный канал над местом сужения расширяется, в последующем возникает застой мочи и сопутствующие расстройства в вышерасположенных органах мочевыделительного тракта. Врожденные и посттравматические передние клапаны анатомически представляют собой дистальную губу широкого дивертикула уретры, вдающуюся в просвет уретрального канала.
Классификация
В зависимости от локализации дефекта клапаны уретры бывают передними, расположенными в пенальном или бульбозном отделах, и задними, перекрывающими просвет простатической части мочеиспускательного канала. Задние клапаны выявляются в 10 раз чаще передних. В соответствии с классификацией различают три типа задних клапанов:
- Клапаны I типа. Выявляются у 90-95% пациентов. В виде парусовидного образования исходят из гребня уретры. Проходят в дистальном направлении, огибая с обеих сторон семенной бугорок, и сливаются перед мембранозным отделом уретрального канала. В тяжелых случаях полностью перекрывают ток мочи.
- Клапаны II типа. Представляют собой естественные складки слизистой, проходящие от семенного бугорка к мочепузырной шейке. Обнаруживаются редко. Поскольку практически никогда не влияют на пассаж мочи по уретре, считаются вариантом анатомической нормы. Могут сочетаться с клапанами I типа.
- Клапаны III типа. Диагностируются в 5-10% случаев аномалии. Представлены сплошной диафрагмой или кольцевидной мембраной с центральным отверстием, расположенными на уровне семенного бугорка или несколько дистальнее по ходу уретры. Могут существенно ограничивать мочевыделение.
Симптомы клапанов уретры
Заболевание обычно проявляется в младенческом возрасте и раннем детстве. У взрослых мужчин с приобретенными клапанными образованиями в передних отделах уретры симптомы обструкции возникают после травмы, диагностического или хирургического вмешательства. В легких случаях наблюдается дизурия, вялая струя мочи, умеренные боли при мочеиспускании, преобладание ночного диуреза. При тяжелом течении со значительной обструкцией уретры в нижних отделах живота пальпируется гипертрофированный мочевой пузырь в виде плотного объемного образования. Реже отмечается ночное и дневное недержание мочи, мочеиспускание учащается до 10-15 раз в сутки, появляется кровь в моче. Лихорадка, боли в пояснице, озноб, слабость выявляются только при присоединении инфекции.
Осложнения
При выраженной обструкции мочеиспускания осложненное течение заболевания наблюдается уже в пренатальном периоде. Возникает маловодие и сочетанная с ним гипоплазия легких, приводящая к легочной и системной гипертензии, а в наиболее тяжелых случаях — перинатальной смерти ребенка. У новорожденных возможно развитие мочевого асцита из-за спонтанной перфорации мочевыводящего тракта. В постнатальном периоде вследствие нарушенного опорожнения мочевого пузыря происходит хроническая задержка мочи, перерастяжение органа, формируется мегауретер, гидроуретеронефроз, пузырно-мочеточниковый рефлюкс. У 44% мальчиков нарушения уродинамики усугубляются за счет присоединения нейрогенной дисфункции мочевого пузыря.
Наличие клапанов уретры способствует размножению патогенной микрофлоры, становящейся причиной уроинфекционных заболеваний. У 15% пациентов возникают циститы, у 89-91% — пиелонефриты, у 4-5% — пионефроз, у 1% — уретриты. В 2-3% случаев на фоне клапанного поражения уретры развивается уросепсис. У 4-4,5% больных диагностируется мочекаменная болезнь, при этом образуются преимущественно мочепузырные камни. У 28-29% пациентов длительное течение заболевания осложняется хронической почечной недостаточностью, формирующейся вследствие уменьшения количества функционирующих нефронов, снижения скорости фильтрации мочи.
Диагностика
Диагностический поиск для выявления или исключения клапанов уретры должен осуществляется врачом-урологом, проводится всем мальчикам с нарушением нормального мочевыделения и ослабленной струей мочи. Современные скрининговые методы позволяют заподозрить наличие дефекта еще на этапе пренатальной УЗИ-диагностики. Для обнаружения в мочеиспускательном канале клапанов назначают такие исследования, как:
- Рентгенография. Уретрография обычно выполняется как продолжение микционной цистографии. Позволяет обнаружить мешкообразное или воронкообразное расширение задней части уретры с резким сужением под ним и нормальным диаметром дистального отдела (симптом «песочных часов» на уровне клапана).
- Эндоскопия. Особенностью аномалии является свободное прохождение эндоскопа через участок сужения. По данным уретроскопии удается точно локализовать клапаны, хорошо заметные в виде гипертрофированных складок слизистой, полулунных образований, диафрагм, мембран.
- Уродинамическое исследование. При проведении урофлоуметрии объемная скорость мочеиспускания снижена. Результаты цистометрии опорожнения подтверждают наличие обструкции, а профилометрия внутриуретрального давления обеспечивает определение места сужения уретрального канала.
Как дополнительный метод рекомендовано бужирование уретры. Головчатый буж легко проходит участок сужения, однако при его извлечении может ощущаться препятствие на уровне клапанов. Вспомогательными критериями диагностики являются данные цистографии – мочевой пузырь на рентгеновских снимках увеличен, его шейка раскрыта. При длительном существовании клапанного дефекта выявляются мочепузырные дивертикулы, признаки мочеточниково-пузырного рефлюкса.
Дифференциальная диагностика проводится с неспецифическими воспалительными заболеваниями (уретритом, циститом, уретеритом, пиелонефритом), стриктурами или полипами уретры, поликистозом почек, хроническим гломерулонефритом. Для оценки состояния верхних отделов мочевыводящей системы назначаются УЗИ почек, мочеточников, мочевого пузыря, экскреторная урография, нефросцинтиграфия, общий и биохимический анализ мочи, нефрологический комплекс. При необходимости по направлению уролога пациента осматривает нефролог, андролог, онколог.
Лечение клапанов уретры
Единственным методом, позволяющим восстановить нормальную проходимость мочеиспускательного канала у больных с клапанным дефектом, является хирургическое вмешательство. Тип и время проведения операции определяются возрастом ребенка, диаметром уретры, наличием сопутствующих расстройств. Рекомендованными методиками являются:
- Эндоуретральная резекция клапанов. Мальчикам старшего возраста и взрослым пациентам осуществляется эндоскопическая операция с использованием электрохирургических инструментов. Абляция клапанов позволяет восстановить самостоятельное мочеиспускание и улучшить уродинамику. Благодаря изобретению детских резектоскопов уменьшенного диаметра появилась возможность проведения вмешательства в более раннем возрасте без риска образования стриктуры в уретральном канале.
- Промежностные операции. Младенцам, диаметр уретры которых недостаточен для введения даже самого маленького резектоскопа, резекция клапанов выполняется через уретростому в области промежности. Поскольку вмешательство производится в непосредственной близости от наружного сфинктера мочеиспускательного канала, что может привести к его повреждению, у грудных детей возможно наложение временной цистостомы с абляцией клапана через несколько месяцев.
Использование эпицистостомы для разрушения уретральных клапанов мало распространено в связи со сложностью визуализации наиболее важного участка клапанных образований I типа — места слияния их створок. При наличии осложнений удаление дефекта является первым этапом хирургического лечения. В последующем могут проводиться операции по наложению уретероцистоанастомоза (при наличии мегауретера или пузырно-мочеточникового рефлюкса), аугментационная цистопластика или кишечная пластика мочевого пузыря (при значительной гипертрофии детрузора). Мальчикам с диагностированной уроинфекцией резекция клапанов откладывается до санации мочевыводящих путей, для облегчения отвода мочи осуществляется цистостомия, уретерокутанеостомия.
Прогноз и профилактика
При своевременной диагностике и раннем хирургическом удалении клапана наблюдается полное выздоровление. При отсутствии адекватного лечения у 17% больных возникает необратимое снижение функции почек, требующее проведения ЗПТ или трансплантации органа. Специфические меры первичной профилактики врожденных клапанов уретры не разработаны. Вторичная профилактика заключается в раннем обнаружении аномалии строения уретры, что позволяет избежать осложнений. Риск образования передних уретральных клапанов после вмешательств на мочеиспускательном канале можно уменьшить за счет тщательного соблюдения техники операции.
Важную роль в снижении частоты дефекта имеет работа женской консультации — выявление беременных из группы риска (наличие экстрагенитальных заболеваний, возраст до 18 или старше 35 лет, осложнения при предыдущих беременностях), регулярная оценка состояния плода. Антенатальная диагностика аномалии позволяет своевременно назначить раннее лечение. При обнаружении с помощью УЗИ-скрининга гидронефроза, расширения чашечно-лоханочной системы, маловодия и других признаков, свидетельствующих о грубом нарушении уродинамики плода, может возникнуть вопрос о прерывании беременности.
Источник
Увеличение размеров мочевого пузыря плода наиболее часто выявляется при аномалиях уретры (атрезия, агенезия, стриктуры, стеноз), синдроме задних уретральных клапанов, синдроме prune belly и мегацистис-микроколон-интестинальном гипоперистальтическом синдроме.
Синдром задних уретральных клапанов (СЗУК) характеризуется нарушением проходимости уретры, что приводит к ретроградным изменениям мочевыделительной системы. На долю СЗУК приходится 38% всех обструктивных уропатий на низком уровне и наблюдается у плодов мужского пола. В крайне редких случаях у плодов женского пола аналогичная эхогра-фическая картина может вызываться агенезией/атрезией уретры.
При СЗУК в ходе ультразвукового исследования плода отмечается стойкая дилатация мочевого пузыря, не проходящая при динамическом наблюдении. Стенки мочевого пузыря утолщены и составляют более 2 мм. В большинстве случаев дилатация мочевого пузыря сочетается с расширением проксимального отделауретры, а при прогрессировании процесса присоединяется и расширение вышерасположенных отделов мочевыделительной системы (мегауретер, гидронефроз), причем процесс носит двусторонний характер. Иногда могут наблюдаться мочевой асцит, уриномы, кальцификация дистрофированных стенок мочевого пузыря]. Маловодие встречается у 50-60% плодов с СЗУК, что значительно ухудшает прогноз, так как приводит к гипоплазии легких.
В последние годы при диагностике обструкции мочевыводящих путей на низком уровне все чаще зарубежными специалистами используется везико-амниотическое шунтирование. Эта процедура, по данным G. Bernaschek и соавт., позволяет добиться выживаемости 70% плодов. Однако авторы отмечают, что в большинстве случаев причина развития обструкции, определяющая прогноз, не может быть установлена пренатально. Прогностическими ультразвуковыми критериями являются количество околоплодных вод, срок выявления выраженного маловодия (длительное маловодие до 28 нед приводит к гипоплазии легких), длительность и тяжесть поражения функции почек. Так как наличие обструктивной уропатии повышает риск выявления хромосомной патологии, то перед установкой шунта необходимо проведение пренатального кариотипирования. Для определения функции почек проводят пункцию увеличенного мочевого пузыря или лоханки с последующим биохимическим анализом полученной мочи. Признаками сохраненной функции почки являются концентрация Na не более 100 ммоль/л, CI – не более 90 ммоль/л, осмолярность – не более 210 ммоль.
Установка пузырно-амниотического шунта должна производиться не позднее 20-22 нед. Однако, несмотря на своевременно и правильно проведенное внутриутробное лечение, все равно остается вероятность неблагоприятного исхода вследствие развития гипоплазии легких.
Для восстановления проходимости уретры в постнатальном периоде новорожденномуустанавливается катетер или проводится везикостомия. После коррекции водного и электролитного баланса и стабилизации функции почек проводятудаление или электрокоагуляцию заднихуретральных клапанов, но даже в случаях успешной хирургической коррекции часть детей погибает вследствие легочной недостаточности. У многих детей и после операции отмечается снижение функции почек, которое может прогрессировать и приводить к потере почки.
Синдром prune belly характеризуется тремя признаками: гипотензией или полным отсутствием мышц передней брюшной стенки, наличием большого атоничного мочевого пузыря (часто сочетается с мегауретером и гидронефрозом) и двусторонним крипторхизмом. Синдром prune belly относится к редким заболеваниям, его частота составляет 1 случай на 35 000 – 50 000 новорожденных.
Клинически условно выделяют три степении тяжести синдрома. Легкая – наличиетреххарактерных основных признаков; при средней, дополнительно к триаде, наблюдается расширение мочеточников; при наиболее тяжелой форме синдрома – гидроуретер, гидронефроз, дисплазия почек, а также возможно развитие гипоплазии легких, скелетных деформаций и характерных особенностей лица из-за выраженного маловодия.
Большинство случаев синдрома является спорадическим. Имеющиеся семейные наблюдения не позволяют однозначно судить о типе наследования синдрома. В литературе сообщалось о сочетании синдрома prune belly с хромосомными аномалиями (трисомия 13, 18 и 45,X).
Ультразвуковая диагностика синдрома prune belly возможна с 14-15 нед беременности. В начале II триместра беременности основным эхографическим признаком является мегацистис, при этом увеличенный мочевой пузырь может занимать большую часть брюшной полости плода. Во II-III триместре беременности эхографическая картина характеризуется наличием резко расширенного, неопорожняющегося мочевого пузыря с гипертрофией его стенок и истончением передней брюшной стенки. При выраженном процессе отмечается присоединение двустороннего мегауретера, гидронефроза и маловодия. В результате прогрессирования патологических изменений может развиться мочевой асцит.
Пренатальное обследование должно включать кариотипирование и тщательное ультразвуковое исследование, включающее подробную оценку анатомии лица и внутренних органов плода. В случае гибели плода или новорожденного показано тщательное патологоанатомическое исследование.
При выявлении синдрома prune belly, сочетающегося с выраженным маловодием, следует предложить прерывание беременности, так как прогноз при таком сочетании крайне неблагоприятный. Уретральная обструкция приводит к развитию выраженного маловодия и как следствие к развитию гипоплазии легких. В 20% случаев наступает антенатальная гибель и 50% детей умирают в первые два года жизни. В случаях легкой степени тяжести синдрома прогноз также в большинстве случаев неблагоприятный, хотя сообщалось о случаях успешной хирургической коррекции. Успешность хирургической коррекции зависит от степени вовлеченности в патологический процесс органов и систем плода.
Внутриутробная декомпрессия мочевого пузыря может предотвратить развитие развернутой картины синдрома. Описаны случаи, когда даже однократной декомпрессии было достаточно для устранения функциональной обструкции. В то же время другие исследователи сообщают о развитии клинической картины синдрома несмотря на проведение ранней декомпрессии.
– Читать далее “Мегацистис-микроколон-интестинальный гипоперистальтический синдром. Диагностика гипоперистальтического синдрома.”
Оглавление темы “Патология половой системы плода.”:
1. Мегацистис у плода. Синдром задних уретральных клапанов плода.
2. Мегацистис-микроколон-интестинальный гипоперистальтический синдром. Диагностика гипоперистальтического синдрома.
3. Экстрофия мочевого пузыря. Диагностика экстрофии мочевого пузыря.
4. Удвоение мочевого пузыря. Диагностика удвоения мочевого пузыря.
5. Определение пола плода. УЗИ диагностика пола плода.
6. Аномалии мужских половых органов плода. Гидроцеле у плода.
7. Гипоспадия. УЗИ диагностика гипоспадии у плода.
8. Аномалии женских половых органов. Кисты яичников у плода.
9. Гидрометрокольпос у плода. Врожденные пороки развития опорно-двигательной системы у плода.
10. Диагностика врожденных пороков развития опорно-двигательной системы у плода. Пренатальная диагностика ВПР ОДС.
Источник
Очень сочувствую вашей золовке, но похоже тут без вариантов – изменения не транзиторные, т.е существуют уже давно и захватили и всю мочевую систему ребенка, и плаценту с оболочками и дальше их количество и выраженность будут нарастать, а срок еще небольшой. Те случаи что хорошо кончаются, о которых мне доводилось читать или были локальное увеличение только пузыря на маленьком сроке, спонтанно проходившее, или более обширный синдром , но уже нед в 28+ впервые выявленный. Тогда долгое хирургическое лечение, возможно раннее родоразрешение и совершенно здоровым увы такой ребенок не будет.
Но конечно против ее воли никто ничего не сделает, хотя прессовать могут. В любом случае надо проверить кариотип, если генетические отклонения эта инфа важна на будущее. И если они есть то пролонгировать б смысл теряется.
2
Автор, к сожалению при таком сочетании диагнозов 50%, что ребенок погибнет внутриутробно и 50%, что умрет в течение первых двух лет жизни.
Как бы страшно это не звучало, но такие диагнозы – это 100% прерывание беременности.
Мне очень жаль, но увы…
1
Очень сочувствую вашей золовке. Диагноз очень серьезный и ситуации заканчиваются по разному. У кого то умирает в утробе у кого-то живёт. В любом случае ей надо самой решать. Но ребенок будет всю жизнь инвалидом, на лекарствах. Надо будет много операций и больших денег. Если она к этому готова, то пусть рожает. Это ее выбор и никто не вправе отправлять ее насильно на аборт. Удачи ей, терпения и счастья!
Ох… Спасибо, девочки. Я не представляю, как буду завтра с ней разговаривать… Что ей говорить? Как поддержать?
узнайте к чему она склоняется. Поговорите с братом и узнайте, что он думаете, расскажите ему о рисках и о предстоящих операциях, лечение. Узнайте, что он хочет. Может он вовсе не готов к такому. Его мнение тоже надо учитывать. Поговорите с золовкой, скажите, что вы узнали от знакомых о таком диагнозе. Спросите, точно ли диагноз подтверждён генетиками, может стоит ещё раз перепроверить или сходить на консультацию к врачам у которых были в практике такие или похожие случаи. И вообще узнайте, что он думает делать.
Они должны принять сами решение и вы можете только пожалеть и поддержать словами или материально к сожалению
И вообще узнайте, что ОнА думает делать*
шансов практически нет… сочувствую.
Тоже сочувствую очень, сама столкнулась с этим год назад на первом скрининге (в 12 недель). Поставили диагноз мегацистис, увеличение мочевого пузыря, расширение почечных лоханок. В медико-генетическом центре был консилиум, решили прерывать. Потом после анализа хориона выяснилось что с генетикой все было в порядке (46ХУ). До сих пор не могу найти причину.