
Синдромами lange nielsen и jervell

Синдром Жервелла и Ланге-Нильсена. Кардиоаудиторный или сурдокардиальный синдромJervell и Lange-Nielsen описали синдром глубокой врожденной нейросенсорной глухоты с патологией ЭКГ, характеризующейся удлинением интервала Q—Т, обморочными приступами и иногда внезапной необъяснимой смертью в детском возрасте. Впоследствии синдром был изучен многими авторами (Lcvine, Woodworth, Fraser, Froggatt, James, Jervell, Thingstad, Endsjo, Lisker, Finkelstein, Kallfelz, Sanchez Cascos et al., van Bruggen et al., Fauchier et al., Athanasiou, Weiner). Fraser, Froggatt и Murphy установили его частоту, составляющую от 3 до 4 случаев на 1 млн. рождений. Кроме того, они определили, что синдром может составлять до 1% среди наследственной глухоты. Однако Sanchez Cascos с сотр. нашли только один случай среди 511 глухонемых детей. Fay с соавт. обнаружили одного больного с этим синдромом среди 1100 глухих детей. Кроме того, 4 больных страдали обмороками без удлинения интервала Q—Т, а у 5 больных отмечалось удлинение интервала Q—Т без обмороков. Орган слуха. У всех больных отмечалась врожденная двусторонняя глубокая нейросенсорная глухота. Лабораторные данные. Изменения ЭКГ характеризовались удлинением интервала Q—Т с широкой T-волной, которая могла быть вертикальной, зазубренной, двухфазной или перевернутой. Степень удлинения интервала Q—Т варьировала как внутри одной семьи, так и между разными семьями, но почти всегда превышала 0,5 с (при максимуме 0,4 с В норме). Так как интервал Q—Т удлиняется в зависимости от скорости сердечных сокращений, существует простая формула для определения его величины (Ljting): Q—T= (RRX0,2) +0,18±0,04. В нескольких случаях была обнаружена легкая гипохромная анемия (Jervell, Langc-Nielsen, Fraser, Froggatt, James, Jervell et al., Lamy et al, Kallfelz). Патология. Описаны многочисленные результаты вскрытия. При макроскопическом и гистологическом исследовании сердца в большей части случаев патологии не обнаружили. Специальное исследование проводящей системы сердца показало выраженное сужение главной ветви артерии синусового узла, в результате чего развивался инфаркт узла (Friedmann et al.). Fraser, Froggatt и James обнаружили, что в сердце этих больных отсутствует нормальная гликогенсодержащая перинуклеарная зона в волокнах Пуркинье.
Изменения височной кости описаны Friedmann с соавт.. Наиболее уникальным изменением являлось накопление ПАСК-положительных глыбок гиалина в атрофичной сосудистой полоске. Отмечалась почти полная дегенерация кортиева органа с утратой чувствительных клеток. Покровная мембрана была сморщенной или втянутой, а рейснерова мембрана была сращена с основной мембраной, практически облитсрируя улитковый ход. Чувствительный эпителий утрикулюса и саккулюса был атрофичным, а гребешок дезорганизованным. Отмечалась умеренная утрата нервных клеток спирального узла. Наследственность. Синдром отчетливо наследуется по аутосомно-рецессивному типу. Часто встречаются кровнородственные браки между родителями (Fraser, Froggatt, Murphy, Lamy et al., Sanchez Cascos et al.). У гетерозигот может наблюдаться умеренное удлинение интервала Q—Т (Fraser, Froggatt и Murphy). Предполагают, что заболевание наследуется сцепленно с Rh-фактором (Friedmann, Fraser, Froggatt). Диагноз. Описаны некоторые изменения ЭКГ (такие, как удлинение интервала Q—Т) без глухоты (Johansson, Jorming, van der Straaten, Bruins, Singer et al.). В этих случаях также наблюдались внезапная смерть и/или приступы потери сознания. Однако патология в этих семьях наследовалась по доминантному типу. Jervell описал еще один доминантно наследующийся синдром множественных экстрасистол с приступами фибрилляции желудочков, но с нормальным интервалом Q—Т. Отмечается определенное сходство настоящего заболевания с изменениями, наблюдающимися при синдроме Рефсума. При обоих синдромах выражены нейросенсорная глухота, нарушения сердечной проводимости с удлинением интервала Q—Т и аномальной Т-волной и иногда наступает внезапная смерть. Однако при синдроме Рефсума глухота выявляется в зрелом возрасте и, кроме того, в сыворотке крови повышен уровень фитановой кислоты. Обморочные приступы при синдроме Ланге-Нильсена могут быть ошибочно диагностированы как эпилептические припадки. Однако ЭЭГ нормальна, в то время как ЭКГ резко изменена. Кроме того, после обморочных состояний у детей не развивается глубокого оглушения. Отмечено несколько атипичных случаев. Mathews с соавт. сообщили об аутосомно-доминантной форме с легкой высокочастотной глухотой. Глухота и сердечная патология могут наследоваться как независимые доминантные признаки. Furlancllo с сотр. и Athanasiou и Muller-Seydlitz также описали доминантную форму заболевания у взрослых с легким дефектом слуха. Предполагают также, что в этих семьях имелся синдром «LEOPARD». Читатель, конечно, помнит, что причиной удлинения интервала Q—Т могут служить: гипокалиемия, гипокальциемия, гипомагниемия и введение квинидина и фенотиазина. Прогноз. С возрастом отмечается незначительное прогрессирование дефекта слуха. У больных может наблюдаться различное число обморочных приступов. Примерно в половине случаев больные погибают к 15-летнему возрасту. Было выявлено несколько больных в возрасте старше 21 года. Выводы. Главными чертами этого синдрома являются: – Также рекомендуем “Ушная-зубная или отодентальная дисплазия. Липодистрофия лица, плеч и кисты костей с глухотой” Оглавление темы “Генетические болезни с глухотой”:
|
Источник
Медицинская статья опубликована в рубрике: Кардиология, ЛОР, СИНДРОМЫ | Декабрь 9th, 2013
 В 1957 г. Жервелл совместно с Ланге Нильсеном описали синдром, характеризующийся постоянным сочетанием врожденной глухоты, с приступами синкопы и характерными электрокардио графическими изменениями.
В 1957 г. Жервелл совместно с Ланге Нильсеном описали синдром, характеризующийся постоянным сочетанием врожденной глухоты, с приступами синкопы и характерными электрокардио графическими изменениями.
Синдром Жервелла Ланге Нильсена — генетическая аномалия с семейным характером, поражающая оба пола.
Этиопатогенез синдрома Жервелла Ланге Нильсена.
Не существует предположения, которое объяснило бы одновременное нарушение слуха и миокарда. Предположения тех, кто придерживается единого мнения, объясняет аномалию сердца гипокалиемией, а глухоту — нарушением кровоснабжения всего слухового аппарата.
Удлинение интервала Q — Т обусловлено нарушением реполяризации (с неуточненной этиологией), с переменчивой интенсивностью от одного дня к другому, определяя такое же переменчивое изменение интервала Q — Т.
Сердечное синкопе объясняется, как приступ типа Адамса-Стокса, или как приступ венечной ишемии. Смерть, наступившая при этих синкопах, может быть вызвана бесконечным удлинением интервала Q—Т, полной предсердно-желудочковой диссоциацией или фибрилляцией желудочков. Следует подчеркнуть, что синкопе появляются все реже и реже, по мере развития ребенка.
Синдром встречается, как у взрослых, так и у детей; по Fraser, представляет приблизительно 1% всех случаев врожденной глухонемоты. Наследственная передача происходит рецессивным образом, вероятно, как результат палеотропического эффекта аномального гена у гомозигот с полным клиническим проявлением; у гетерозигот синдром проявляется только удлинением интервала Q—T.
Симптоматология синдрома Жервелла Ланге Нильсена.
Врожденная глухота — всегда присутствует; она двусторонняя, обусловливая глухонемоту маленького ребенка. Глухота перцепторного типа, не затрагивает низкие звуки.
Приступы синкопе — повторные, вызываемые обыкновенно волнениями, холодом или даже без видимой причины, продолжаются несколько секунд и внезапно прекращаются.
Во время синкопы у больных отмечается:
- бледность;
- тахикардия;
- полипное;
- больной стонет, теряет или нет сознание;
- иногда появляются судороги.
Несколько минут после прекращения синкопе, у больного затуманенное сознание, затем постепенно приходит в нормальное состояние.
Диагностика синдрома Жервелла Ланге Нильсена.
Электрокардиографические характерные изменения заключаются в:
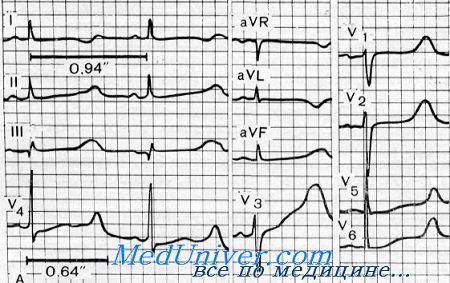
— патологическом удлинении интервала Q—Т (представляя постоянные и значительные электрокардиографические изменения). В сравнении с нормальным пределом этого интервала (0,31 секунды) величины, встречающиеся у больных, всегда намного выше (в среднем они колеблются между 0,40 и 0,55 секунд при сердечном ритме выше 90/мин).
Фонокардиограмма показывает, что длительность электрической систолы всегда больше механической:
- изменения волны Т очень разнообразна: отрицательная волна, положительная и двухфазная;
- комплекс QRS и интервал Р — Q — всегда нормальные.
Непостоянно добавляется и гипохромная анемия, умеренной интенсивности.
Течение и прогноз синдрома Жервелла Ланге Нильсена.
Течение, как и дальнейший прогноз, неблагоприятные. Смерть ребенка может произойти во время приступа синкопе, тем более, что часто приступ синкопе диагностируется, как приступ эпилепсии и лечат его, как таковой, лишая, таким образом, больного соответствующей экстренной терапии.
Лечение синдрома Жервелла Ланге Нильсена
а) Лечебное:
- экстренное назначение (если диагноз был уточнен) дигиталиса в 1% растворе, 5—6 капель в день, 5 дней в неделю (с еженедельным 2-х дневным перерывом во избежание накопления дигиталиса в миокарде), в течение 3—4 недель. Применением дигиталиса можно избежать смерти во время синкоп, укорачивая и нормализируя интервал Q — R, в отличие от хинидина, который способствует его удлинению (отсюда вытекает абсолютное противопоказание хинидина при приступах синкопе);
- хлористый калий в дозе от 0,5—1 г/день перорально;
- успокаивающие (диазепам).
б) Профилактическое:
- электрокардиографическое исследование всех новорожденных и грудных детей, страдающих судорогами и состоянием синкопе;
- внимание при дифференциальном диагнозе приступа синкопе, который ошибочно может быть принят за приступ эпилепсии; от своевременного и правильного диагноза зависит жизнь больного.
Источник
Синдром удлиненного интервала QT на ЭКГ
• Для синдрома удлиненного интервала QT характерны 2 признака: удлинение интервала QT (длительность расчетного интервала QT превышает 0,44 с) и желудочковая тахикардия с обмороками.
• В дополнение к этим признакам отмечаются высокая волна U, уплощенный или отрицательный зубец Т, а также синусовая тахикардия.
• Врожденная форма данного синдрома встречается реже и является генетически гетерогенным заболеванием, приобретенная форма часто бывает обусловлена антиаритмической терапией.
• Врожденную форму синдрома удлиненного интервала QT лечат блокаторами бета-адренергических рецепторов, а при отсутствии эффекта от медикаментозной терапии при необходимости имплантируют кардиовертер/дефибрил-лятор. При приобретенной форме следует, прежде всего, отменить препараты, которые могли стать причиной удлинения интервала QT.
Синдром удлиненного интервала QT (синоним: синдром QT) делят на врожденную, генетически гетерогенную, форму и приобретенную, или медикаментозную, форму. Врожденная форма встречается крайне редко (1 случай на 10 000 новорожденных). Клиническое значение синдрома QT состоит в том, что как врожденная, так и приобретенная его форма проявляется желудочковой тахикардией.
I. Врожденный синдром удлиненного интервала QT (синдромы Джервелла-Ланге-Нильсена и Романо-Уорда)
В патогенезе врожденного синдрома QT играют роль мутации генов, кодирующих белки ионных каналов, приводящие к недостаточной активности калиевых каналов или повышенной активности натриевых каналов. Синдром удлиненного интервала QT может проявляться в форме синдрома Джервелла-Ланге-Нильсена и синдрома Романо-Уорда.
Характерными признаками синдрома Джервелла-Ланге-Нильсена являются:
• удлинение интервала QT
• глухонемота
• эпизоды обмороков и внезапная смерть.
При синдроме Романо-Уорда глухонемоты нет.
Первые клинические проявления врожденного синдрома QT появляются уже в детском возрасте. Характерны повторные эпизоды обмороков, появляющиеся на фоне симпатикотонии, например, когда ребенок плачет, испытывает стресс или кричит.
К важнейшим ЭКГ-признакам синдрома QT относятся:
• удлинение интервала QT, т.е. длительность расчетного интервала QT превышает 0,44 с (в норме он равен 0,35-0,44 с)
• желудочковая тахикардия (пируэтная тахикардия: быстрая и полиморфная форма)
• синусовая брадикардия в покое и при нагрузке
• уплощенный или отрицательный зубец Т
• высокая или двухфазная волна U и слияние зубца Т и волны U
• зависимость продолжительности интервала QT от ЧСС
При измерении интервала QT следует проявить внимательность, чтобы не включить в интервал волну U (корригированный интервал QT; интервал QTC по Базетту). Относительный интервал QT (например, по Лепешкину или Хегглину и Гольцману) измерить проще, но значение его при этом менее точное. В норме оно составляет 100±10%.
При синдроме QT отмечается неравномерное удлинение фазы реполяризации, что облегчает механизм повторного входа волны возбуждения, способствуя появлению желудочковой тахикардии (torsade de pointes, пируэтная тахикардия) и фибрилляции желудочков.
Лечат синдром QT блокаторами бета-адренергических рецепторов, а в случае резистентности к этим препаратам имплантируют кардиовертер/дефибриллятор.
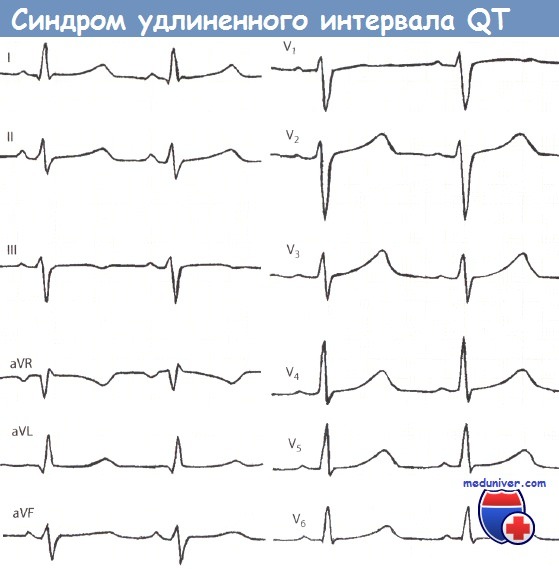
Синдром удлиненного интервала QT (синдром Романо-Уорда).
ЧСС 90 ударов в минуту, длительность QT 0,42 с, относительная длительность интервала QT составляет 128%, откорректированный интервал QTC удлинен и равен 0,49 с.
II. Приобретенный синдром удлиненного интервала QT
Причины, вызывающие приобретенный синдром удлиненного интервала QT, могут быть разные. Ниже перечислены только имеющие наибольшее клиническое значение:
• антиаритмические препараты (например, хинидин, соталол, амиодарон, аймалин, флекаинид)
• нарушение электролитного баланса (например, гипокалиемия)
• блокада ножки ПГ и уширение комплекса QRS
• гипотиреоз
• ИБС
• антибиотикотерапия (например, эритромицином)
• злоупотребление алкоголем
• миокардит
• церебральное кровоизлияние
В типичных случаях приобретенный синдром QT бывает связан с приемом антиаритмических препаратов, особенно хинидина и соталола. Клиническое значение данного синдрома велико, учитывая, что, как и при врожденной форме, приобретенный синдром QT сопровождают приступы желудочковой тахикардии.
Частота возникновения приступов желудочковой тахикардии у больных с приобретенным синдромом удлиненного интервала QT составляет 2-5%. Типичным примером являются так называемые хинидиновые обмороки. Изменения на ЭКГ такие же, как при врожденном синдроме QT.
Лечение подразумевает, прежде всего, отмену «причинного» препарата и введение, помимо прочего, раствора лидокаина.
Особенности ЭКГ при синдроме удлиненного интервала QT:
• Изменение интервала QT (в норме интервал QTC <0,44 с)
• Склонность к желудочковой тахикардии
• Врожденная форма: при обмороках некоторым больным показана имплантация кардиовертера/дефибриллятора
• Приобретенная форма: отмена антиаритмических препаратов (частая причина синдрома)
Учебное видео оценки комплекса QRS на ЭКГ в норме и при патологии
– Вернуться в оглавление раздела “Кардиология.”
Оглавление темы “Расшифровка ЭКГ (электрокардиограммы)”:
- Расшифровка холтеровского мониторинга электрокардиограммы (ЭКГ)
- Признаки перикардита на ЭКГ
- Признаки миокардита на ЭКГ
- Признаки хронического легочного сердца на ЭКГ
- Признаки ТЭЛА (тромбоэмболии легочной артерии, острого легочного сердца) на ЭКГ
- Признаки синдрома Вольфа-Паркинсона-Уайта (WPW) на ЭКГ
- Классификация синдрома Вольфа-Паркинсона-Уайта (WPW): типы А и В
- АВ-узловая пароксизмальная тахикардия при синдроме Вольфа-Паркинсона-Уайта (WPW)
- Признаки гипокалиемии на ЭКГ
- Синдром удлиненного интервала QT на ЭКГ
Источник
Рубрика МКБ-10: I45.8
МКБ-10 / I00-I99 КЛАСС IX Болезни системы кровообращения / I30-I52 Другие болезни сердца / I45 Другие нарушения проводимости
Определение и общие сведения[править]
Наследственный синдром удлинения интервала QT
Синонимы: врожденный синдром длительного интервала QT
Наследственный синдром удлинения интервала QT является генетическим заболеванием сердца, которое характеризуется удлинением интервала QT на ЭКГ и высоким риском развития угрожающих жизни аритмий.
Распространенность синдрома оценивается близко к 1 в 2500 родившихся живыми.
Этиология и патогенез[править]
Генетическая основа заболевания была выявлена в середине девяностых и опеределены все каузальные гены LQTS, которые кодируют субъединицы кардиальных ионных каналов или белки, участвующие в модуляции ионных потоков. Мутации в этих генах (KCNQ1, KCNH2, KCNE1, KCNE2, CACNA1C, CAV3, SCN5A, SCN4B) вызывают удлинение продолжительности потенциала действия. Наиболее распространен вариант, который вызывается мутациями гена KCNQ1.
Клинические проявления[править]
Два основых проявления врожденного синдрома длительного интервала QT: обморочные эпизоды, которые могут приводить к остановке сердца и внезапной смерти, а также электрокардиографические признаки, включая удлинение интервала QT и аномалии Т-волн.
Другие уточненные нарушения проводимости: Диагностика[править]
Учитывая характерные особенности LQTS, типичные случаи не представляют диагностические трудности для врачей , осведомленных о болезни. Тем не менее, пограничные случаи являются более сложными и требуют оценки различных электрокардиографических, клинических и семейных признаков. Кроме того, молекулярный скрининг теперь является частью диагностического процесса.
Дифференциальный диагноз[править]
Другие уточненные нарушения проводимости: Лечение[править]
Лечение всегда следует начинать с бета-блокаторов, если нет к ним противопоказаний. Если у пациента возникает обморочные эпизод на фоне бета-блокаторов – должны быть выполнена симпатическая денервация левых отделов сердца. Имплантируемый кардиовертер-дефибриллятор следует рассматривать как окончательное терапевтическое решение.
Прогноз
Прогноз, как правило, хороший у пациентов. Тем не менее, есть несколько тяжелых исключений: больные с синдромом Тимоти (характеризуется выраженной QT пролонгации интервала, 2:1 функциональной атриовентрикулярной блокадой и синдактилией), синдромом Джервелла и Ланге-Нильсена с мутацией KCNQ1 (тяжелая форма, врожденная глухота и очень раннее возникновение сердечной аритмии) и синдромом Романо-Уорда (2:1 атриовентрикулярная блокада и очень раннее возникновение аритмии).
Профилактика[править]
Прочее[править]
Семейное прогрессирующее нарушение проводимости сердца
Синонимы: семейная болезнь Ленегра, семейная болезнь Лева, семейная болезнь Лева-Ленегра,
семейная прогрессирующая блокада сердца, дефект ножки пучка Гиса
Определение и общие сведения
Семейное прогрессирующее нарушение проводимости сердца является наследственным расстройством проводимости, которое может прогрессировать вплоть до полной атриовентрикулярной блокады сердца. Семейная болезнь Лева-Ленегра может протекать бессимптомно или проявляться одышкой, головокружением, приступами потери сознания, болью в животе, развитием сердечной недостаточности или внезапной смертью.
На сегодняшний день в литературе описано более 50 случаев патологии. Семейное прогрессирующее нарушение проводимости сердца наследуется по аутосомно-доминантному типу с неполной пенетрантностью и вариабельной экспрессивностью. Рецессивные или спорадические формы встречаются редко.
Этиология и патогенез
Семейное прогрессирующее нарушение проводимости сердца представляет собой дегенеративный процесс, поражающий проводящие пути сердца комплекса пучок Гиса-волокна Пуркинье. Мутации в трех генах идентифицированы в качестве каузальных – SCN5A, SCN1B и TRPM4. Мутации в генах NKX2, TBX5, PRKAG2 и LMNA были также выявлены у пациентов, у которых семейное прогрессирующее нарушение проводимости сердца сопровождается наличием врожденного порока сердца. Изолированное семейное прогрессирующее нарушение проводимости также было обнаружено в семьях с носительством мутаций в одном из этих генов. Ген GJA5 был ассоциирован с ранним началом и тяжелым течением патологии, был описан у двух кровных родственников.
Клинические проявления
Первые проявления семейной болезни Лева-Ленегра возникают в разном возрасте. Семейное прогрессирующее нарушение проводимости может протекать бессимптомно или проявляется одышкой, головокружением, приступами потери сознания, болью в животе, развитием сердечной недостаточности, а также внезапной смертью при развитии полной сердечной блокады. Описаны случаи синкопы на фоне физической нагрузки. Возможно прогрессирование заболевания от нормальной ЭКГ-картины до блокады правой ножки пучка Гиса и вплоть до полной блокады сердца.
Диагностика
Диагностика семейной болезни Лева-Ленегра основана на наличии в семейном анамнезе синкопальных состояний, имплантации электрокардиостимулятора и случаев внезапной смерти, а также на результатах электрокардиографии, свидетельствующих о наличии выраженных нарушений проводимости – полной блокады правой или левой ножек пучка Гиса, блокады передней или задней ветви левой ножки пучка Гиса, удлинении интервала PR или полной АВ-блокады с широкими комплексами QRS. В большинстве случаев обнаруживаются нормальные строение и сократительная функция сердца, однако иногда полная АВ-блокада может привести к дилатации левого желудочка и развитию сердечной недостаточности. Желудочковая тахикардия или тахикардия по типу «пируэт» могут быть зарегистрированы на ЭКГ в фазу восстановления после проведения теста с физической нагрузкой или на фоне полной АВ- блокады. С помощь эхокардиографии или МРТ выявляются врожденные пороки сердца или кардиомиопатия. При обнаружении изолированного прогрессирующего нарушения проводимости сердца у пациента в молодом возрасте – необходимо проведение скрининга на наличие генов, отвественных за развитие наследственной патологии.
Дифференциальный диагноз
Дифференциальный диагноз включает синдром Бругада, идиопатическую фибрилляцию желудочков, синдром удлинения QT, волчанку новорожденных, прогрессирующую семейную блокаду II типа и синдром внезапной смерти ребенка.
Лечение
Лечение семейной болезни Лева-Ленегра включает своевременную имплантацию постоянного искусственного водителя ритма. Больных с блокадами любой степени рекомендуется обследовать в динамике каждые 6 месяцев, а членам их семей с нормальными результатами ЭКГ рекомендуется обследование минимум один раз в год. Необходимо ограничить применение препаратов, замедляющих проводимость. У пациентов с мутациями гена SCN5A следует проводить превентивное лечение лихорадки, которая является провоцирующим фактором. При установлении диагноза необходимо обследование родственников первой линии.
Прогноз
Для пациентов с семейной болезнью Лева-Ленегра отсутствует стратификация риска по генотипу. Отмечается высокая частота развития внезапной смерти у пациентов с АВ-блокадой I степени в комбинации с двухпучковой блокадой или у больных с выраженной симптоматической АВ-блокадой. У пациентов, которым была выполнена имплантация искусственного водителя ритма, прогноз является благоприятным и практически соответствует таковому в общей популяции, за исключением больных с мутациями в гене LMNA, которые могут демонстрировать желудочковую тахикардию и внезапную кардиальную смерть. Таким пациентам показана имплантация кардиовертера-дефибриллятора в случае тяжелых нарушений проводимости.
Синдром Тимоти
Синдром Тимоти представляет собой мультисистемное расстройство. Характеризуется сердечными и неврологическими особенностями, а также аномалиями конечностей и лица, которые включают удлинение интервала QT, синдактилию пальцев кистей и стопы, сплющенную переносицу, низкорасположенные уши, маленькую верхнюю челюсть, тонкую верхнюю губу и аутизм или проявления спектра аутистического расстройства.
Синдром Джервелла и Ланге-Нильсена
Синонимы: синдром удлинение интервала QT-глухота
Синдром Джервелла и Ланге-Нильсена – аутосомно-рецессивный вариант наследственного синдрома удлинения интервала QT, характеризующийся сочетанием врожденной глубокой двусторонней потерей слуха, длительным интервалом QT на ЭКГ и желудочковыми тахиаритмиями.
Синдром Джервелла и Ланге-Нильсена встречается очень редко. Распространенность неизвестна, оценки варьируют от 1/200 000 до 1/1 000 000, но чаще встречается в регионах с близкородственными браками.
Синдром Джервелла и Ланге-Нильсена вызван гомозиготными или компацндными гетерозиготными мутациями либо в гене KCNQ1 (локус LQT1, 11p15.5), либо в гене KCNE1 (локус LQT5, 21q22.1-q22.2).
Синдром Романо-Уорда
Синонимы: синдром удлиненного QT Романо-Уорда
Синдром Романо-Уорда – аутосомно-доминантный вариант наследственного синдрома удлинения интервала QT, характеризующийся синкопальными эпизодами и электрокардиографическими аномалиями (удлинение QT, аномалии Т-волн и желудочковая тахикардия по типу пируэта).
Распространенность синдрома удлиненного QT Романо-Уорда оценивается в 1/2 500. Синдром Романо-Уорда наследуется аутосомно-доминантно с низкой пенетрантностью.
Синдром Романо-Уорда может быть результатом мутаций в генах, кодирующих субъединицы ионных каналов миокарда (KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2 и SCN4B) или белков, взаимодействующих с ионными каналами миокарда (ANK2, CAV3, AKAP9 или SNTA1).
Источники (ссылки)[править]
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник