
Синдромы с х сцепленной хромосомой

Х-сцепленное наследование: принципы, примеры
Описано более 400 заболеваний, при которых патологический рецессивный ген расположен на Х-хромосоме.
Примеры Х-сцепленных рецессивных заболеваний:
• Дальтонизм (неразличение красного и зелёного цветов).
• Миодистрофии Дюшенна и Беккера.
• Синдром ломкой Х-хромосомы.
• Недостаточность Г-6-ФД.
• Синдром Гунтера (мукополисахаридоз II).
• Гемофилия А и В.
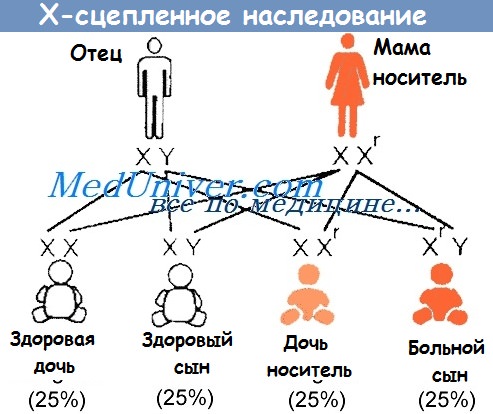
При Х-сцепленном рецессивном наследовании:
• поражаются мужчины;
• женщины могут быть носителями, но обычно здоровы;
• иногда у женщины-носителя есть мягкие симптомы заболевания (манифестирующий носитель);
• у каждого сына женщины-носителя риск заболевания 1:2 (50%);
• у каждой дочери женщины-носителя риск носительства 1:2 (50%);
• все дочери больных мужчин будут носителями;
• сыновья больных мужчин будут здоровы, поскольку мужчина передаёт своему сыну Y-хромосому.
Семейный анамнез может быть отрицательным, поскольку новые мутации и мозаицизм гонад встречаются сравнительно часто. Выявление в семье женщин-носительниц требует обследования детей на предмет мягких клинических проявлений и проведения специфических биохимических или молекулярных анализов. Выявление носителей важно, потому что у женщин-носительниц риск рождения больного сына составляет 50% независимо от того, кто будет его отцом.
Кроме того, Х-сцепленные рецессивные заболевания проявляются зачастую очень тяжело.
При Х-сцепленном рецессивном наследовании:
• Поражаются мужчины; женщины могут быть носителями, но обычно клинически здоровы или имеют мягкие проявления болезни.
• Семейный анамнез может быть отрицательным — в связи с новыми мутациями и мозаицизмом гонад.
• Выявление женщин-носителей важно для проведения генетического консультирования.
Наследование по Х-сцепленному доминантному типу. Х-сцепленные заболевания с доминантной мутацией редки. Поражаются мужчины и женщины, например при форме витамин D-резистентного рахита. При некоторых заболеваниях среди пациентов мужского пола существуют летальные исходы, и на обследование попадают только пациентки, например при синдроме Ретта и недержании пигмента.
Наследование по Y-сцепленному типу. Y-сцепленные заболевания чрезвычайно редки. Наследование по Y-сцепленному типу может проявляться только у пациентов мужского пола с передачей от больного отца всем его сыновьям. Y-сцепленные гены определяют половую принадлежность и сперматогенез, поэтому такие мутации часто связаны с бесплодием.
– Также рекомендуем “Синдром ломкой Х-хромосомы: принципы, примеры”
Оглавление темы “Генетические болезни детей”:
- Синдром Тернера: причины, клиника
- Синдром Клайнфелтера. Реципрокные транслокации и делеции
- Аутосомно-доминантное наследование: принципы, примеры
- Аутосомно-рецессивное наследование. Близкородственный брак
- Х-сцепленное наследование: принципы, примеры
- Синдром ломкой Х-хромосомы: принципы, примеры
- Митохондриальное и цитоплазматическое наследование. Импринтинг и дисомия от одного родителя
- Полигенное или мультифакторное наследование: принципы, примеры
- Анализ ДНК детей. Методы
- Пороки развития детей: патогенез, классификация
Источник
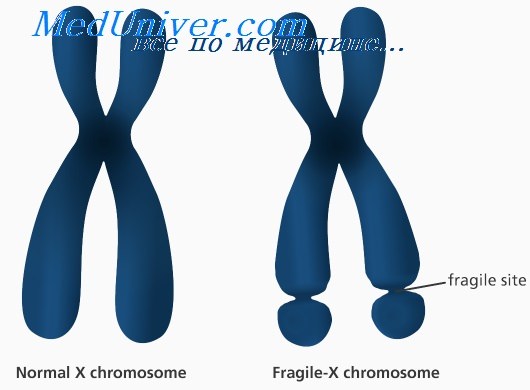
Синдром ломкой Х-хромосомы: причины, диагностика, лечениеЭтиология и встречаемость синдрома ломкой Х-хромосомы. Синдром ломкой Х-хромосомы (MIM №309550) — Х-сцепленное заболевание с задержкой умственного развития, вызванное мутациями в гене FMR1 в Xq27.3. Синдром ломкой Х-хромосомы встречается с частотой 16-25 на 100 000 в общей популяции среди мужчин и в два раза реже среди женщин. Синдром ломкой Х-хромосомы составляет 3-6% всех случаев умственной отсталости среди мальчиков с положительным семейным анамнезом по умственной отсталости при отсутствии врожденных пороков. Патогенез синдрома ломкой Х-хромосомыПродукт гена FMR1, FMRP, экспрессируется во многих типах клеток, но наиболее сильно в нейронах. FMRP может сопровождать определенный подкласс мРНК от ядра к рибосомам. Более 99% мутаций в гене FMR1 — экспансия нуклеотидного повтора (CGG)n в 5′-нетранслируемом участке гена. В нормальных аллелях FMR1 число повторов CGG составляет от 6 до приблизительно 50. В патогенных аллелях (или при полных мутациях) количество повторов более 200. Аллели с более чем 200 повторами CGG обычно имеют гиперметилированную последовательность повторов CGG и смежного промотора FMR1. Гиперметилирование инактивирует промотор FMR1, вызывая снижение экспрессии FMRP. Полные мутации возникают из аллелей премутации (от 59 до 200 повторов CGG) с передачей мутантного аллеля FMR1 от матери (но не от отца); фактически при отцовской передаче премутации часто, наоборот, сокращаются. Полные мутации не могут возникать из нормальных аллелей. Поскольку длина неустойчивых повторов CGG увеличивается в каждом последующем поколении, если они передаются женщиной, обычно наблюдается увеличение числа пораженных потомков в последующих поколениях в семье; этот феномен называется генетической антиципацией. Риск экспансии премутации в полную мутацию возрастает с увеличением числа повторов в премутации. Тем не менее не все премутации одинаково предрасположены к экспансии. Хотя премутации встречаются сравнительно часто, переход в полную мутацию наблюдают только в ограниченном количестве гаплотипов, т.е. когда есть склонность гаплотипа к экспансии. Эта склонность гаплотипа частично может быть связана с присутствием нескольких триплетов AGG, вставленных в последовательность повторов CGG; оказывается, такие триплеты AGG тормозят экспансию повторов CGG, следовательно, их отсутствие в некоторых гаплотипах может предрасполагать к экспансии.
Фенотип и развитие синдрома ломкой Х-хромосомыСиндром ломкой Х-хромосомы вызывает умеренную умственную отсталость у мужчин и легкую умственную задержку у женщин. Наиболее пораженные индивидуумы также имеют поведенческие аномалии, включая гиперактивность, размахивание руками, истерики, плохой зрительный контакт и признаки аутизма. Физические характеристики мужчин изменяются с пубертатом. До полового созревания пораженные мальчики имеют несколько увеличенный размер головы и некоторые другие неотчетливые симптомы; после наступления половой зрелости у них частые более отчетливые признаки (длинное лицо с выдающейся челюстью и лбом, крупные ушные раковины, макроорхидизм). Поскольку эти клинические признаки не уникальны для синдрома ломкой Х-хромосомы, диагноз зависит от молекулярного обнаружения мутаций. Пациенты с синдромом ломкой Х-хромосомы имеют нормальную продолжительность жизни. Почти все мужчины и 40-50% женщин, унаследовавших полную мутацию, будут иметь синдром ломкой Х-хромосомы. Тяжесть фенотипа зависит от мозаицизма метилирования повторов и их числа. Поскольку полные мутации неустойчивы, некоторые пациенты имеют смесь клеток с числом повторов, колеблющимся от премутации до полной мутации (мозаицизм числа повторов). Все мужчины с мозаицизмом числа повторов больны, но часто имеют более высокие показатели умственного развития, чем пациенты с полной мутацией в каждой клетке; у женщин с мозаицизмом числа повторов клинические проявления варьируют от нормы до полного проявления. Аналогично некоторые пациенты имеют смесь клеток с метилированием повторов CGG и без него (мозаицизм метилирования повторов). Все мужчины с мозаицизмом метилирования больны, но часто имеют более высокие показатели умственного развития, чем с гиперметилированием в каждой клетке; женщины с мозаицизмом метилирования также могут быть здоровыми или больными.
Очень редко пациенты имеют полную мутацию, неметилированную во всех клетках; независимо от пола, степень тяжести у них варьирует от нормы до полной клиники. Кроме того, у женщин фенотип зависит от степени смещения инактивации Х-хромосомы. Носительницы премутации (но не полных мутаций) имеют 20% риск ранней дисфункции яичников. Мужчины-носители премутации имеют риск развития синдрома FXTAS. FXTAS проявляет себя как поздняя прогрессирующая мозжечковая атаксия с интенционным тремором. У больных могут также присутствовать снижение краткосрочной памяти и двигательных функций, когнитивные нарушения, а также паркинсонизм, периферическая нейропатия, проксимальная мышечная слабость нижних конечности и дизавтономия. Пенетрантность FXTAS зависит от возраста, обнаруживается в 17% в течение шестого десятилетия жизни, в 38% в течение седьмого десятилетия, в 47% в течение восьмого десятилетия и в трех четвертях старше 80 лет. FXTAS может встречаться и у некоторых женщин — носительниц премутации. Особенности фенотипических проявлений синдрома ломкой Х-хромосомы: Лечение синдрома ломкой Х-хромосомыК настоящему времени никакого патогенетического лечения при синдроме ломкой Х-хромосомы нет. Помощь направлена на обучение и фармакологическое лечение поведенческих проблем. Риски наследования синдрома ломкой Х-хромосомыРиск того, что женщина с премутацией будет иметь больного ребенка, определяется размером премутации, полом плода и семейным анамнезом. Эмпирически риск для носителя перестройки иметь больного ребенка может достигать 50% для каждого мальчика и 25% для каждой девочки, но зависит от размера премутации. На основе анализа сравнительно небольшого количества матерей-носительниц известно, что риск повторения может снижаться, если премутация уменьшается со 100 до 59 повторов. Пренатальная диагностика доступна за счет использования ДНК плода из ворсин хориона или амниоцитов. Пример синдрома ломкой Х-хромосомы. Р.Л., 7-летний мальчик, направлен в клинику педиатрии в связи с умственной задержкой и гиперактивностью. Он не смог посещать детский сад, поскольку был агрессивным, не в состоянии выполнять задания, имел бедные речевые и двигательные навыки. Несмотря на задержанное развитие, он не потерял основных этапов: сидел к 10-11 мес, ходить начал в 20 мес, говорил два или три ясных слова в 24 мес. В остальном ребенок здоров. Его мать и тетя по матери имели небольшие проблемы обучения в детстве, дядя по матери умственно задержан. Данные медицинского осмотра в норме, за исключением гиперактивности. Врач рекомендовал несколько тестов, включая кариотипирование, функциональные исследования щитовидной железы и ДНК-анализ на синдром ломкой Х-хромосомы. Анализ гена FMR1 методом блот-гибридизации по Саузерну соответствовал синдрому ломкой Х-хромосомы. – Также рекомендуем “Недостаточность глюкозо-6-фосфат дегидрогеназы (Г6ФД): причины, диагностика, лечение” Оглавление темы “Генетические болезни”:
|
Источник
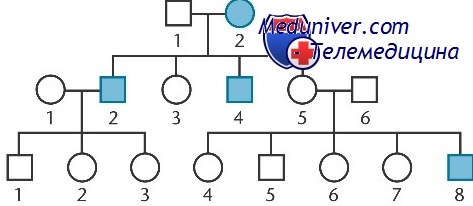
Характеристика Х-сцепленного рецессивного наследования. ПримерыНаследование рецессивных Х-сцепленных фенотипов вызывает отчетливо выраженные и легко узнаваемые типы родословных. Х-сцепленная рецессивная мутация обычно фенотипически проявляется у всех имеющих ее мужчин и только у гомозиготных женщин. Следовательно, Х-сцепленное рецессивное заболевание обычно ограничено мужчинами и редко бывает среди женщин (см. раздел о манифестных гетерозиготах далее в этой главе). Гемофилия А — классическое Х-сцепленное заболевание свертывания крови, вызванное недостаточностью фактора VIII, одного из белков, участвующих в свертывании крови. Наследственная природа гемофилии и даже тип передачи известны с древних времени, заболевание стало известно как «королевская гемофилия» из-за наличия среди потомков королевы Великобритании Виктории, которая была носительницей. Как уже сказано, Xh — мутантный аллель фактора VIII, вызывающий гемофилию А, Хн — нормальный аллель. Если больной гемофилией женится на здоровой женщине, все сыновья получают отцовскую хромосому Y и материнскую X и здоровы, все дочери получают отцовскую Х-хромосому с алле-лем гемофилии и становятся облигатными носителями. Гемофилия больного дедушки, не проявившаяся у его собственных детей, имеет 50% вероятность появления у сыновей любой из его дочерей. В то же время она не проявится среди потомков его сыновей. Дочь женщины-носителя имеет 50% шанс оказаться носительницей. Случайным образом, прежде чем проявиться в потомке-мужчине, Х-сцепленный рецессивный аллель может передаваться необнаруженным через серию женщин-носительниц. Гомозиготные больные женщиныГен для Х-сцепленного заболевания может случайно присутствовать как у отца, так и у материносительницы, и тогда девочки могут оказаться гомозиготными больными, как показано на родословной Х-сцепленного дальтонизма, сравнительно частого Х-сцепленного заболевания. Большинство Х-сцепленных болезней достаточно редкие, поэтому женщины нечасто бывают гомозиготными, если ее родители не кровные родственники.
Манифестные гетерозиготы и несбалансированная инактивация при Х-сцепленных болезняхВ тех редких случаях, когда женщина-носительница рецессивного Х-сцепленного аллеля имеет фенотипические проявления болезни, ее называют манифестной гетерозиготой. Манифестные гетерозиготы описаны для многих Х-сцепленных рецессивных заболеваний, включая дальтонизм, гемофилию А (классическая гемофилия, недостаток фактора VIII), гемофилию В (болезнь Кристмаса, недостаток фактора IX), мышечную дистрофию Дюшенна, синдром Вискотта-Олдрича (Х-сцепленный иммунодефицит) и несколько Х-сцепленных заболеваний глаз. Будет ли гетерозиготная женщина манифестной, зависит от множества факторов. Во-первых, поскольку Х-инактивация происходит случайно, но на этапе эмбрионального развития, когда эмбрион имеет менее 100 клеток, соотношение в различных тканях женщин-носительниц клеток с нормальным и мутантным аллелем в активной хромосоме может сильно варьировать. Если случится, что патологический аллель чаще присутствует на активной, а нормальный на неактивной хромосоме, появляется несбалансированный или «скошенный» результат Х-инактивации. Если такая «скошенная» инактивация представлена в соответствующих тканях, она может вызвать у женщины-носительницы признаки и симптомы заболевания. Во-вторых, в зависимости от заболевания, о котором идет речь, женщины-гетерозиготы могут иметь весьма разную степень пенетрантности и экспрессивности болезни, даже при равной степени перекоса инактивации, из-за особенностей физиологического функционирования гена. Например, при лизосомной болезни накопления, вызываемой недостаточностью сульфоидуронат сульфатазы (синдром Хантера), те клетки, в которых активна Х-хромосома, несущая нормальный ген, могут передать фермент во внеклеточное пространство, откуда он попадает в клетки с мутантным аллелем и скорректировать дефект. В результате пенетрантность синдрома Хантера среди женских гетерози-гот чрезвычайно низкая, даже когда Х-инактивация значительно отклоняется от ожидаемого случайного отношения 50%-50%. С другой стороны, почти половина всех женщин-гетерозигот по синдрому ломкой Х-хромосомы имеют аномалии развития, хотя обычно меньшей выраженности, чем у мужчин. Кроме манифестных гетерозигот, возможен характерный для нескольких Х-сцепленных заболеваний противоположный вариант несбалансированной или скошенной инактивации (т.е. с мутантным аллелем, преимущественно обнаруживаемым на неактивной Х-хромосоме в некоторых или всех тканях гетерозиготной женщины). В основном такой перекос инактивации наблюдают у бессимптомных гетерозигот. Полагают, что он отражает способность к выживанию или недостаток пролиферативной активности для клеток, первоначально имевших мутантный аллель на активной Х-хромосоме. Феномен скошенной инактивации в соответствующих тканях используют для диагностики состояния носительства для некоторых Х-сцепленных заболеваний, включая некоторые Х-сцепленные иммунодефициты, врожденный дискератоз (Х-сцепленная форма болезни кожи и костного мозга) и недержание пигмента (Х-сцепленное заболевание кожи и зубов). Характеристика рецессивного Х-сцепленного наследования: – Также рекомендуем “Характеристика Х-сцепленного доминантного наследования. Примеры” Оглавление темы “Генетика”:
|
Источник
Здравствуйте. Сегодня я расскажу вам о синдроме ломкой Х-хромосомы и его влиянии на женскую репродуктивную функцию и здоровье потомства, о связи генов и бесплодия, о методах диагностики при раннем истощении резерва яичников. Всеми этими проблемами мы занимаемся в Центре иммунологии и репродукции.
Синдром ломкой X-хромосомы – наследственное заболевание, при котором нарушена работа гена FMR1. Суть генетического нарушения в увеличении количества повторов сочетания из трех «букв» генетического кода CGG. У здоровых людей таких повторов в X-хромосоме не более 50–60. При синдроме ломкой X-хромосомы – более 200.
Состояние, когда повторов от 60 до 200, называется премутацией. У ее носителя практически нет симптомов, но повышен риск возникновения полноценной мутации у потомства. Другие названия патологии – синдром фрагильной хромосомы X и синдром Мартина-Белл.
Мутация в гене FMR1 – одна из самых распространенных вместе с синдромом Дауна причин наследственного интеллектуального нарушения. Но этот генетический дефект привлекает и пристальное внимание врачей-репродуктологов, потому что он приводит к преждевременному истощению овариального резерва и может передаваться ребенку.
Распространенность синдрома ломкой X-хромосомы встречается у одного из 4000 мужчин и одной из 8000 женщин. Такое различие связано с тем, что у женщин две X-хромосомы. Если в одной возникает дефект, вторая может его компенсировать. У мужчин только одна X-хромосома. Поэтому, если им достается мутация FMR1, она проявляется в полную силу.
Премутация встречается очень часто: у каждой из 250 женщин в одной из X-хромосом имеется более 50 повторов CGG.
Почему возникают симптомы?
При экстремальном увеличении количества копий последовательности CGG происходит метилирование промотора – участка ДНК, который активирует экспрессию т.е.преобразование генетического кода в структуру белка гена FMR1. В итоге ген не работает. В организме он отвечает за формирование связей в нервной системе и некоторые другие важные процессы. В итоге возникают нарушения, которые и проявляются характерными симптомами.
Симптомы синдрома Мартина-Белл
Несмотря на то, что премутация у женщин зачастую не проявляется какими-либо симптомами, она нарушает репродуктивную функцию и может привести к проблемам со здоровьем у потомства.
- Во- первых, первичная недостаточность яичников.
У женщин с дефектным геном FMR1 рано происходит истощение овариального резерва, и менопауза наступает до 40 лет. Поэтому при преждевременной менопаузе врач может порекомендовать пройти генетический анализ. По статистике, в 5% случаев раннее истощение овариального резерва становится следствием синдрома ломкой X-хромосомы.
Недостаточность яичников возникает примерно у 25% носительниц премутации.
- Во-вторых, передача дефектного гена потомству.
Если у женщины премутация, ее детям может достаться полноценная мутация, когда число повторов CGG увеличено более 200. При этом у девочки симптомы возникнут с вероятностью 30–50%, а у мальчика почти гарантированно. Мужчина со 100% вероятностью передаст ломкую X-хромосому всем дочерям, но ни одному из сыновей , потому что дочь в любом случае получит одну X-хромосому от отца, а сын – всего одну только от матери.
- Существуют и другие симптомы. В первую очередь синдром ломкой X-хромосомы грозит потомству нарушением интеллекта.
Развивается умственная отсталость легкой или средней степени тяжести. У некоторых детей возникают симптомы аутизма: они много раз повторяют одни и те же слова, фразы, действия, не входят в зрительный контакт во время общения, избегают социума.
- Синдром фрагильной хромосомы X может привести к тремору/атаксии, ассоциированному с ломкой X-хромосомой .
Чаще всего патология поражает мужчин и встречается примерно у трети носителей премутации. Симптомы этого состояния: тремор , мозжечковая атаксия, то есть нарушение чувства равновесия, паркинсонизм, деменция .
Обычно первые признаки появляются после 50 лет, до этого возраста человек чувствует себя абсолютно здоровым.
- Другие возможные проявления синдрома Мартина-Белл.
Характерные черты лица: выступающий лоб и подбородок, большие уши. Высокое сводчатое нёбо. Чрезмерная подвижность суставов. Пролапс митрального клапана и другие проблемы с сердцем.
Сразу после рождения установить диагноз невозможно. Симптомы ломкой X-хромосомы могут появиться только после того, как ребенку исполнится год.
- Диагностика синдрома ломкой Х-хромосомы
Раньше это состояние выявляли с помощью цитогенетических исследований. У человека получали образец клеток, вводили в них фолиевую кислоту, и после этого становилось видно, что в X-хромосоме есть как бы разрыв. Один участок «отшнуровывается». На самом деле хромосома цела, просто такой эффект создается из-за мутации. В настоящее время от цитогенетического анализа отказались, так как он недостаточно точен.
Кстати, раньше синдром Мартина-Белл считался хромосомным заболеванием, как и синдром Дауна. Эта группа патологий связана с нарушением структуры и количества хромосом. В настоящее время известно, что это генная болезнь, то есть нарушение происходит на уровне одного определенного гена.
Современный метод диагностики ломкой хромосомы X – полимеразная цепная реакция , когда с помощью фермента получают много копий ДНК, и конечные продукты выявляют методом капиллярного электрофореза. Это позволяет точно определить мутантный ген и его размер до 200 повторов CGG.
Кому показан анализ на ломкую х-хромосому?
Женщине стоит пройти этот анализ в следующих случаях:
- При ранней до 40 лет менопаузе.
- При нарушении репродуктивной функции, в сочетании с повышенным уровнем фолликулостимулирующего гормона ,пониженном антимюллеровом гормоне , урежении ,то есть более 35 дней в цикле и укорочении – менее 3 дней менструации.
- Если у родственников диагностирован синдром ломкой X-хромосомы.
Во многих странах сейчас исследование на мутацию гена FMR1 у плода включено в обязательную программу скрининга во время беременности. Нарушение намного проще выявить у будущей мамы, чем у ребенка, особенно если у женщины имеется премутация, а ребенку досталась полноценная мутация.
ПЦР хуже выявляет дефекты в гене FMR1, если число копий CGG более 200, можно получить ложноотрицательный результат.
Что делать, если анализ обнаружил это нарушение?
Положительный результат анализа на ломкую Х-хромосому
Если у будущей мамы еще до беременности диагностирована премутация или мутация, это дает шанс не допустить передачи заболевания потомству. В ходе ЭКО врачи могут отобрать эмбрионы с нормальной X-хромосомой.
Если из-за синдрома ломкой X-хромосомы у женщины происходит преждевременное истощение овариального резерва, также может помочь ЭКО. Это состояние обязательно учитывается в ходе процедуры, потому что такие женщины хуже реагируют на гормональную стимуляцию яичников.
Такой анализ можно сделать в Лаборатории ЦИР. Мы проводим все доступные лабораторные исследования, которые помогают разобраться в нарушении репродуктивной функции, оценить риски для потомства, вероятность развития тех или иных осложнений во время беременности.
Если у Вас есть вопросы по теме видео, пишите их в комментариях или приходите на консультацию. Часто доступна онлайн-поддержка, о которой Вы можете узнать по ссылке на нашем сайте.
Источник