
Скрининг на адреногенитальный синдром и муковисцидоз
Скрининг на муковисцидоз. Пренатальная диагностика
В настоящее время невозможно объяснить высокую частоту мутантных аллелей CFTR, наблюдаемую в европеоидных популяциях и составляющую 1 на 50. Болезнь значительно реже встречается в других популяциях, хотя описана у американских индейцев, афроамериканцев и монголоидов (например, среди жителей Гавайских островов азиатского происхождения с частотой приблизительно 1 на 90 000).
Аллель F508 — единственный общий фактически для всех европеоидных популяций из обнаруженных до настоящего времени. Анализ гаплотипов европеоидных популяций указывает, что аллель F508, вероятно, имеет единственное начало. Частота этого аллеля среди всех мутантных аллелей значительно изменяется в разных европейских популяциях, с 88% в Дании до 45% в южной Италии.
В популяциях, в которых частота аллеля F508 составляет приблизительно 70%, около 50% пациентов — гомозиготны по аллелю F508; еще 40% — генетические компаунды F508 и другого мутантного аллеля. Кроме того, приблизительно 70% носителей муковисцидоза имеют мутацию F508.
За исключением F508, остальные мутации в локусе CFTR встречаются редко, хотя в специфических популяциях некоторые аллели могут встречаться чаще других.
Популяционный скрининг на муковисцидоз
В настоящее время муковисцидоз соответствует большинству критериев для программы скрининга новорожденных, за исключением того, что пока не ясно, насколько ранняя идентификация больных новорожденных улучшает долгосрочный прогноз.
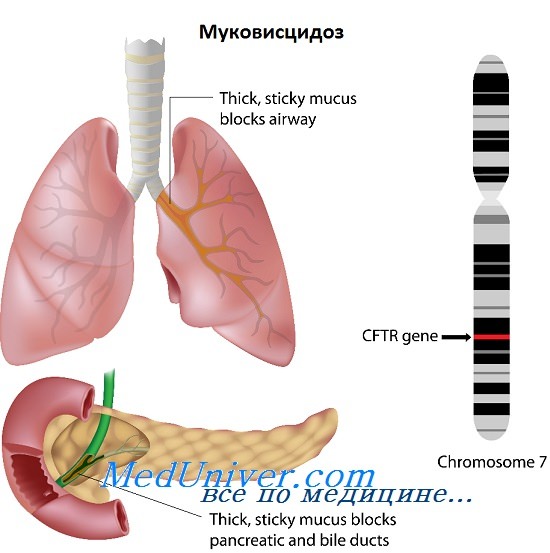
Тем не менее преимущества ранней диагностики (например, улучшенное питание с обеспечением ферментами поджелудочной железы) позволили осуществлять программы неонатального скрининга. Хотя обычно считают, что скрининг носительства нецелесообразен до тех пор, пока не будут обнаруживаться по крайней мере 90% мутаций (в настоящее время — почти 85%), в Соединенных Штатах в течение нескольких лет осуществляют скрининг среди семейных пар на уровне частных медицинских методов.
Генетический анализ семей пациентов и пренатальная диагностика муковисцидоза
Высокая частота аллеля F508 информативна, когда ДНК диагностику проводят пациенту без семейной истории муковисцидоза. Для того чтобы определить статус членов семьи для подтверждения болезни (например, у новорожденного или сибса с неоднозначной симптоматикой), диагностировать носительство или провести пренатальную диагностику можно, используя идентификацию аллеля AF508 в комбинации с панелью из 22 менее частых, но и не редких мутаций, предлагаемую Американским колледжем медицинской генетики.
При условии хорошего знания мутаций муковисцидоза в популяции прямое обнаружение мутации — метод выбора для генетического анализа. Если при отсутствии известной специфической мутации использовать анализ сцепления, точная диагностика возможна фактически во всех семьях.
При беременности с риском 1 к 4 метод выбора — пренатальная ДНК-диагностика на сроке от 10 до 12 нед в тканях, получаемых при БВХ. Биохимические методы пренатальной диагностики, основанные на измерении пищеварительных ферментов (например, щелочной фосфатазы) в амниотической жидкости, имеют высокие ложноположительные показатели, и их больше не используют.
Молекулярная генетика и лечение муковисцидоза. В настоящее время лечение муковисцидоза направлено на борьбу с легочной инфекцией и улучшение питания. Повышение знаний о молекулярном патогенезе может сделать возможным разработку фармакологических вмешательств с целью непосредственно скорректировать аномальный биохимический фенотип.
Кроме того, при муковисцидозе может оказаться возможной генотерапия, но пока есть множество трудностей.
– Также рекомендуем “Генетика мышечных дистрофий Дюшенна и Беккера. Наследование, молекулярные основы”
Оглавление темы “Генетика заболеваний”:
- Генетика семейной гиперхолестеринемии. Наследование, молекулярные основы
- Генетика муковисцидоза. Наследование
- Скрининг на муковисцидоз. Пренатальная диагностика
- Генетика мышечных дистрофий Дюшенна и Беккера. Наследование, молекулярные основы
- Пренатальная диагностика мышечных дистрофий Дюшенна и Беккера. Носительство
- Генетика несовершенного остеогенеза. Наследование
- Генетика болезни Альцгеймера. Наследование, молекулярные основы
- Митохондриальный геном. Наследование
- Мутации в митохондриальной ДНК (мтДНК). Митохондриальные болезни
- Проявления митохондриальных болезней. Фенотипы
Источник
Первые попытки проведения неонатального скрининга (НС) на муковисцидоз (МВ) в Европе предпринимались еще в начале 1970–х годов и сводились к определению содержания альбумина в меконии. И лишь обнаруженное в 1979 г. повышение уровня иммунореактивного трипсина (ИРТ) в плазме крови новорожденных с МВ послужило толчком к началу массового скрининга новорожденных на данное заболевание [1]. Дальнейшее усовершенствование программы НС стало возможным после клонирования в 1989 г. гена CFTR и последующей идентификации специфических CFTR–мутаций в общей популяции, что позволило включить анализ ДНК в скрининговые протоколы [2]. Ежегодно по программе НС в Европе обследовалось более 1,6 млн новорожденных и выявлялось более 400 больных детей. По данным за 2008 г., количество детей, прошедших скрининг, превысило 3 млн в год в связи с внедрением НС на МВ в Великобритании и России. Программа НС оправданна как с медицинской, так и с экономической точки зрения. Ранняя диагностика МВ дает возможность своевременно начать адекватную терапию, что ведет к значительному улучшению качества и продолжительности жизни больных. Кроме того, проведение НС и установка CFTR–генотипа новорожденных с МВ предполагает возможность более раннего генетического консультирования, что может повлиять на репродуктивное поведение супругов и их родственников [3].
В настоящее время в Европе насчитывается около 26 вариантов программ НС, включающих от 2 до 4 последовательных этапов обследования (табл. 1). Первым этапом во всех протоколах является определение уровня ИРТ в высушенном пятне крови новорожденного в первую неделю жизни: весьма чувствительный (85–90%), но не специфичный признак. По данным Европейского консенсуса, гипертрипсинемия в неонатальном периоде встречается при перинатальном стрессе, коньюгационной желтухе новорожденных, при трисомиях 13 [4] и 18 [5] хромосом, у детей с врожденными инфекциями, почечной недостаточностью и атрезией тонкого кишечника, а также в случае нефрогенного несахарного диабета [3]. Популяционное распределение концентраций ИРТ в крови в период новорожденности несколько выше у детей североафриканского происхождения [6] и у афроамериканцев [7], чем у детей из Северной Европы. Поэтому, необходим второй этап обследования.
Использование подхода с определением ИРТ/ДНК в многонациональном обществе не позволяет выявить пациентов с мутациями, специфичными для некоторых этнических групп. В Европейском исследовании мутаций у пациентов с МВ североафриканского и у пациентов турецкого происхождения при использовании стандартных мутационных панелей выявлено только 50% мутаций [3,8]. Это представляет проблему для стран и/или больших городов с многочисленными этническими группами. Некоторые современные программы НС, основанные на определении ИРТ–ДНК, пытаются компенсировать это, сохраняя второй образец ИРТ у детей, у которых мутация CFTR не была обнаружена, однако уровень ИРТ в первом образце был очень высокий [3].
Изучается использование ассоциированного с панкреатитом белка (PAP) в качестве теста 2–го уровня, либо в комбинации с определением ИРТ в рамках теста 1–го уровня. Этот подход позволит избежать проблем, возникающих при анализе CFTR–мутаций, или необходимости повторного забора крови. Разработан комбинированный набор для оценки ИРТ + РАР и планируется проведение пилотных исследований в Нидерландах, Германии и Франции (Jeannette Dankert–Roelse, Olaf Sommerburg и Jacques Sarles, личные сообщения) [3].
Все вышеперечисленные программы могут и должны комбинироваться и проводиться у родственников, идентифицированных скринингом, как в семьях, имеющих больных МВ, так и в популяции в целом (каскадный скрининг). Так как сибсы (братья и сестры больных) имеют шанс в 50% быть носителями (а тети и дяди – в 25%), то этот метод каскадного скрининга может быть эффективным и связанным с минимальными расходами. НС имеет и ряд негативных аспектов. Момент, когда родители впервые слышат о положительном результате скрининга у их ребенка, может быть критическим временем как с медицинской, так и с психологической точки зрения. Основной целью предоставления информации в этот период является обеспечение своевременного проведения предполагаемой медицинской помощи и подтверждающей потовой пробы, если только у ребенка не обнаружена гомозиготная или смешанная гетерозиготная мутация, вызывающая МВ, свидетельствующая о несомненном диагнозе. Однако во время этого периода «максимальной неопределенности» вполне понятно, что тревога родителей может быть значительной. Минимизация интервала между первичным обсуждением результата скрининга на МВ и диагностическим подтверждением благоприятно отражается на психологическом состоянии, а также начале медицинской помощи и развитии взаимного доверия между семьей больного и работниками здравоохранения [2,3,9].
Как указано в Европейском консенсусе, целью НС на МВ является выявление как можно большей доли пациентов с МВ с минимальным количеством ложноположительных результатов по доступной цене [3]. Это может быть достигнуто путем использования различных протоколов скрининга. Поскольку приоритеты скрининга новорожденных во многих странах и регионах различаются в отношении финансирования, удобства забора образцов крови, легкости доступа к клиническим службам и распространенности CFTR–мутации, достичь полного согласования протоколов невозможно. Выбор стратегии зависит от популяционной генетики, стоимости, акцента на определенных целях: максимальной чувствительности, минимальной необходимости или отсутствии необходимости повторного забора образцов крови, частоте ненужного выявления носительства и снижении количества потовых проб. Центральным звеном успеха скрининга новорожденных на МВ является эффективное общение между работниками здравоохранения и родителями. Стандарт общения должен охватывать прескрининговое информирование семей, а также информацию для родителей детей с положительным результатом скрининга новорожденных, новорожденных с МВ и носителей [3,10,11].
С 2006 г. в ряде регионов, а с 1 января 2007 г. во всех субъектах РФ массовый скрининг новорожденных на муковисцидоз был включен в перечень наследственных заболеваний, подлежащих обязательному НС наряду с фенилкетонурией, галактоземией, гипотиреозом и адреногенитальным синдромом в рамках национального приоритетного проекта «Здоровье». Протокол скрининга включает 4 этапа [10,12]: ИРТ, ИРТ2, потовый тест и ДНК–диагностику, – причем только первые три являются обязательными (табл. 2).
Генетическое обследование в РФ проводится только в ряде регионов. Доступность его ограничена высокой стоимостью анализа (например, 3500 рублей за 26 мутаций гена МВТР, что составляет 70–75% от общего числа мутантных аллелей гена МВТР, встречающихся у больных МВ России).
Потовая проба – «золотой стандарт» протокола скрининга на МВ. Согласно рекомендации Ассоциации клинических биохимиков Великобритании, в каждом центре, отвечающем за диагностику МВ, должно проводиться минимум 50 потовых проб в год [3]. В настоящее время в большинстве европейских центров продолжают измерять концентрацию хлоридов в поте (прямой классический биохимический метод Гибсона–Кука). В РФ зарегистрированы и успешно применяются две системы для анализа проводимости пота (непрямое определение хлоридов). Система для сбора и анализа пота Macroduct в комплексе с потовым анализатором Sweat–Chek фирмы Вескор (США) позволяет провести потовую пробу вне лабораторных условий, время сбора пота составляет 30 мин., успешно применяется у детей с первых месяцев жизни. Специально для обследования новорожденных компанией Вескор был разработан аппарат Nanoduct, объединяющий в себе систему для стимуляции потоотделения путем электрофореза 0,1% пилокарпина и анализатор проводимости пота. Благодаря минимальному количеству необходимой для теста потовой жидкости (всего 3–6 мкл) этот аппарат незаменим при обследовании новорожденных в рамках массового скрининга. Важно помнить, что проводимость пота определяется совокупностью всех ионов, присутствующих в потовой жидкости (калий, натрий, хлор, бикарбонат, аммоний и др.), и полученный результат превышает истинную концентрацию хлоридов примерно на 15–20 ммоль/л. Таким образом, положительными считаются результаты выше 80 ммоль/л, а показатели 60–80 ммоль/л – пограничными (табл. 3) [10,12].
Важным достижением практического здравоохранения является централизованная закупка аппаратов потового анализатора Nanoduct МЗ СР РФ для всех субъектов РФ. Специалисты из регионов были обучены работе на аппаратах в Российском центре муковисцидоза.
По данным МЗ и СР РФ, с 1 января 2007 по 31 декабря 2009 г. в РФ на МВ было обследовано 4 160 021 новорожденных. По данным, полученным из всех регионов РФ, выявлено 416 случаев МВ. Предварительная частота заболевания по России составляет 1:10 000 новорожденных. Следует отметить, что не всем детям с повторными высокими значениями ИРТ проводятся потовые пробы, т.к. по разным причинам родители отказываются от данного исследования (до 25% по разным регионам). Таким образом, истинная частота МВ в России значительно выше указанного значения. С учетом данных, полученных из разных субъектов РФ, можно утверждать, что и эта частота значительно варьирует по регионам (табл. 4).
Новорожденные с установленным диагнозом регулярно наблюдаются специалистами Центра МВ: каждые 2 нед. до 3 мес. жизни ребенка, ежемесячно до полугода, каждые 2 мес. с полугода до 1 года и далее ежеквартально (табл. 5). Особенно важно ежемесячное динамическое наблюдение за пациентами без клинических проявлений – массо–ростовые показатели, результаты копрологического исследования (не менее 1 раза/мес. до 1 года), показатели панкреатической эластазы в стуле (2 раза за первый год жизни), рост микрофлоры в посеве мазка из ротоглотки и клинический анализ крови (1 раз в 3 мес.). В случае развития обострения бронхолегочного процесса или отсутствия желаемого контроля над симптомами заболевания может потребоваться более глубокое обследование (рентгенографическое исследование легких или компьютерная томография, липидограмма кала, биохимический анализ крови, протеинограмма и др.) [10].
Лечение ребенка, больного МВ, нужно начинать незамедлительно с момента постановки диагноза. Объем терапии зависит от клинических проявлений и результатов лабораторных и инструментальных методов обследования. У 90% больных МВ первые клинические проявления возникают на первом году жизни и, как правило, в первые месяцы. Всем новорожденным и детям первых месяцев жизни с МВ показано раннее начало кинезитерапии, независимо от наличия у них признаков бронхо–легочного поражения. У грудных детей применяется пассивная техника кинезитерапии, включающая терапевтические положения, контактное дыхание, легкую вибрацию, поглаживания, а также занятия на мяче. На этом этапе очень важен тесный контакт с ребенком, все занятия должны быть приятны малышу. У детей с малейшими симптомами бронхиальной обструкции кинезитерапия применяется в комплексе с муколитическими препаратами и бронходилататорами.
По данным Verhaeghe C. с соавт. из Бельгии, в легочной ткани плодов с МВ отмечено достоверное повышение уровня провоспалительных белков, что говорит о раннем начале воспалительных процессов, предшествующих развитию инфекции. Именно поэтому, на наш взгляд, оправданно раннее назначение дорназы альфа (Пульмозим, «Ф. Хоффманн–Ля Рош Лтд.») в связи с наличием у этого препарата наряду с хорошим муколитическим эффектом противовоспалительного действия, характеризующегося снижением в бронхоальвеолярной жидкости маркеров воспаления (нейтрофильная эластаза, ИЛ–8) [10,12].
Всем новорожденным с МВ, имеющим клинические проявления кишечного синдрома или низкие показатели фекальной эластазы–1 (активность может меняться в течение первого года жизни), показана заместительная терапия микросферическими панкреатическими ферментами под контролем копрограммы, частоты и характера стула, ежемесячной прибавки веса. Обязательным является назначение жирорастворимых витаминов [10,12].
В настоящее время в Московском центре МВ наблюдается 42 ребенка, больных МВ, выявленных по программе НС с июня 2006 г. по март 2010–го (табл. 6).
В 2009 г. с целью определения эффективности НС на МВ нами был проведен анализ CFTR–мутаций в высушенных пятнах крови 990 новорожденных с первым положительным тестом на ИРТ, родившихся в 2008 г. в г. Москве и составивших группу риска на МВ. В ходе исследования CFTR–мутации были обнаружены у 47 индивидов. При этом число мутантных аллелей CFTR гена составило 53, или 2,7%, и было представлено следующими CFTR–мутациями: F508del – обнаружена в 28 случаях (68%), CFTRdele 2,3 (21kb) – в 7 (17%) случаях, 2184insA – в 2 (5%) случаях, 3821delT – 1 (4%) , L138insA – 2 (4%), 2143delT – в 1 (2%) образце ДНК.
В ходе исследования была выявлена девочка 1 года 4 мес., с генотипом CFTRdele2,3(21kb)/CFTRdele2,3 (21kb), которая по НС не вошла в группу риска (ИРТ I – 236 нг/мл, ИРТ II – 12нг/мл) и не была своевременно диагностирована. Семья ребенка была приглашена на консультацию в центр МВ. Результат потового теста – 112 ммоль/л. На момент осмотра массо–ростовые показатели ребенка соответствовали возрастной норме, в анамнезе – неоднократные ОРВИ, жирный стул, госпитализация с подозрением на острую кишечную непроходимость.
Таким образом, мы установили, что частота мутантных аллелей гена у новорожденных группы риска на МВ (с первым положительным ИРТ) составляет 0,02575 (0,02017
÷0,03241), что значительно выше частоты данных мутаций в российской популяции (0,00642). Возможно, имеет место влияние гетерозиготного носительства мутаций F508del, CFTRdele2,3 (21kb), 3821delT, L138insA, 2143delT, 2184insA в гене CFTR, выявленных в ходе исследования, на повышение уровня ИРТ, как следствие функциональной недостаточности поджелудочной железы у новорожденных.
Доля ложноотрицательных результатов НС на МВ в 2008 г. составила 0,1%, что не противоречит общеевропейским данным.
Большинство специалистов в области МВ приходят к выводу, что НС на МВ оправдан, во–первых, с экономической точки зрения, так как позволяет предотвратить рождение больных МВ, в семьях, где уже есть больной ребенок, и способствует появлению в этих семьях здоровых детей; во–вторых, с медицинских позиций, так как продолжительность жизни больных, выявленных с помощью скрининга, выше, чем в других группах [3,13]. Кроме того, скрининг сокращает время подчас мучительной постановки диагноза.
Европейской ассоциацией МВ создана рабочая группа по неонатальному скринингу, в 2007 г. в нее включены представители из России. Основной задачей группы является анализ данных разных стран и регионов Европы, что, в свою очередь, может способствовать в будущем оптимизации программ по скринингу.
Значение скрининга на МВ в России в целом может быть оценено только через несколько лет, при условии регулярного финансирования программы. Кроме того, для ощутимых результатов, сопоставимых с европейскими или американскими, необходимо понимание государством важности не только своевременного выявления больных МВ, но и создания необходимых условий для их наблюдения и лечения.
Литература
1. Crossley JR, Elliott RB, Smith PA. Dried–blood spot screening for cystic fibrosis in the newborn. Lancet 1979;1(8114):472–4.
2. Southern KW, Munck A, Pollit R, Castellani C et al. A survey of newborn screening for cystic fibrosis in Europe. J of Cystic Fibrosis 2007; 6:57–65.
3. Castellani C, Southern KW, Brownlee K. et al. European best practice guidelines for cystic fibrosis neonatal screening. Journal of Cystic Fibrosis Volume 8 (2009) 153–173.
4. Priest FJ, Nevin NC. False positive results with immunoreactive trypsinogen screening for cystic fibrosis owing to trisomy 13. J Med Genet 1991;28:575–6.
5. Heeley AF, Fagan DG. Trisomy 18, cystic fibrosis, and blood immunoreactive trypsin. Lancet 1984;1:169–70.
6. Cheillan D, Vercherat M, Chevalier–Porst F, Charcosset M, Rolland MO, Dorche C. False–positive results in neonatal screening for cystic fibrosis based on a three–stage protocol (IRT/DNA/IRT): should we adjust IRT cut–off to ethnic origin? J Inherit Metab Disease 2005;28:813–8.
7. Giusti R. New York State Cystic Fibrosis Newborn Screening Consortium. Elevated IRT levels in African–American infants: implications for newborn screening in an ethnically diverse population. Pediatr Pulmonol 2008;43: 638–41.
8. Lakeman P, Gille JJP, Dankert–Roelse JE, et al. CFTR mutations in Turkish and North African cystic fibrosis patients in Europe: implications for screening. Genetic Testing 2008;12:25–35.
9. Kharrazi M, Kharrazi LD. Delayed diagnosis of cystic fibrosis and the family perspective. J Pediatr 2005;147:S21–5.
10. Толстова В.Д., Каширская Н.Ю., Капранов Н.И. Массовый скрининг новорожденных на муковисцидоз в России. Фарматека. – 2008. – №1. – С.1–5.
11. Comeau AM, Parad RB, Dorkin HL et al. Population–based newborn screening for genetic disorders when multiple mutation DNA testing is incorporated: a CF newborn screening model demonstrating increased sensitivity but more carrier detections. Pediatrics 2004;113(6):1573–81.
12. Муковисцидоз. Современные достижения и актуальные проблемы. Методические рекомендации. Издание третье (первое 2001) переработанное и дополненное. Под редакцией Капранова Н.И., Каширской Н.Ю. М.: ООО «4ТЕ Арт». 2008. – с.124.
13. Brice P, Jarrett J, Mugford M. Genetic screening for cystic fibrosis: An overview of the science and the economics. J of Cystic Fibrosis 2007; 6:255–261.
Источник