
Спинальная амиотрофия верднига гоффмана код мкб

Связанные заболевания и их лечение
Описания заболеваний
Стандарты мед. помощи
Содержание
- Синонимы диагноза
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Другие названия и синонимы
Детская спинальная мышечная атрофия, Спинальная мышечная атрофия I типа.
Названия
Название: Амиотрофия Верднига-Гоффмана.
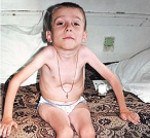
Амиотрофия Верднига-Гоффмана
Синонимы диагноза
Детская спинальная мышечная атрофия, Спинальная мышечная атрофия I типа.
Описание
Амиотрофия Верднига. Гоффмана – наиболее злокачественная спинальная мышечная атрофия, развивающаяся с рождения или в первые 1 – 1,5 года жизни ребенка. Характеризуется нарастающими диффузными мышечными атрофиями, сопровождающимися вялыми парезами, прогрессирующими до полной плегии. Как правило, амиотрофия Верднига-Гоффмана сочетается с костными деформациями и врожденными аномалиями развития. Диагностическую основу составляет анамнез, неврологический осмотр, электрофизиологические и томографические исследования, анализ ДНК и изучение морфологического строения мышечной ткани. Лечение слабо эффективно, направлено на оптимизацию трофики нервной и мышечной тканей.
Амиотрофия Верднига-Гоффмана
Дополнительные факты
Амиотрофия Верднига-Гоффмана является самым тяжелым вариантом из всех спинальных мышечных атрофий (СМА). Ее распространенность находится на уровне 1 случай на 6-10 тыс. Новорожденных. Носителем измененного гена, обуславливающего возникновение заболевания, является каждый 50-й человек. Но благодаря аутосомно-рецессивному типу наследования, патология у ребенка проявляется только тогда, когда соответствующая генетическая аберрация имеется и у матери, и у отца. Вероятность рождения ребенка с патологией в такой ситуации составляет 25%.
Амиотрофия Верднига-Гоффмана имеет несколько форм: врожденную, промежуточную (раннюю детскую) и позднюю. Целый ряд специалистов выделяет последнюю форму как самостоятельную нозологию — амиотрофию Кугельберга-Веландера. Отсутствие этиотропного и патогенетического лечения, ранний летальный исход ставят курирование пациентов с болезнью Верднига-Гоффмана в ряд наиболее сложных задач, стоящих перед современной неврологией и педиатрией.
Причины
Амиотрофия Верднига-Гоффмана — наследственная патология, кодируемая поломкой в генетическом аппарате на уровне локуса 5q13 5-й хромосомы. Ген, в котором происходят мутации, получил название survival motor neuron gene (SMN) — ген, ответственный за выживание мотонейронов. У 95% пациентов с болезнью Верднига-Гоффмана отмечается делеция теломерной копии этого гена. Тяжесть СМА прямо коррелирует с протяженностью участка делеции и сопутствующим наличием изменений (рекомбинации) в генах H4F5, NAIP, GTF2H2.
Результатом аберрации SMN-гена является недоразвитие мотонейронов спинного мозга, локализующихся в его передних рогах. Следствием становится недостаточная иннервация мышц, приводящая к их выраженной атрофии с потерей мышечной силы и прогрессирующим угасанием способности совершать активные двигательные акты. Основную опасность представляет слабость мышц грудной клетки, без участия которых невозможны движения, обеспечивающие дыхательную функцию. При этом сенсорная сфера на всем протяжении заболевания остается интактной.
Симптомы
Врожденная форма (СМА I) клинически манифестирует до 6-месячного возраста. Внутриутробно может проявляться вялым шевелением плода. Зачастую мышечная гипотония отмечается с первых дней жизни и сопровождается угасанием глубоких рефлексов. Дети слабо кричат, плохо сосут, не могут держать голову. В отдельных случаях (при более позднем дебюте симптомов) ребенок учится держать голову и даже сидеть, но на фоне развития заболевания эти навыки быстро исчезают. Характерны ранние бульбарные нарушения, понижение глоточного рефлекса, фасцикулярные подергивания языка.
Данная амиотрофия Верднига-Гоффмана сочетается с олигофренией и нарушениями формирования костно-суставного аппарата: деформациями грудной клетки (воронкообразной и килевидной грудной клеткой), искривлением позвоночника (сколиозом), контрактурами суставов. У многих пациентов выявляются другие врожденные аномалии: гемангиомы, гидроцефалия, косолапость, дисплазия тазобедренных суставов, крипторхизм и пр.
Течение СМА I наиболее злокачественное с быстро нарастающей обездвиженностью и парезом дыхательной мускулатуры. Последний обуславливает развитие и прогрессирование дыхательной недостаточности, выступающей основной причиной летального исхода. В связи с нарушением глотания возможен заброс пищи в дыхательные пути с развитием аспирационной пневмонии, которая может явиться смертельно опасным осложнением спинальной амиотрофии.
Ранняя детская форма (СМА II) дебютирует после 6. Месячного возраста. К этому периоду дети имеют удовлетворительное физическое и нервно-психическое развитие, в соответствии с возрастными нормами приобретают навыки держать голову, переворачиваться, садиться, стоять. Но в подавляющем большинстве клинических случаев дети так и не успевают научиться ходить. Обычно эта амиотрофия Верднига-Гоффмана манифестирует после перенесенной ребенком пищевой токсикоинфекции или другого острого инфекционного заболевания.
В начальном периоде периферические парезы возникают в нижних конечностях. Затем они достаточно быстро распространяются на верхние конечности и мускулатуру туловища. Развивается диффузная мышечная гипотония, происходит угасание глубоких рефлексов. Наблюдаются контрактуры сухожилий, тремор пальцев, непроизвольные мышечные сокращения (фасцикуляции) языка. На поздних стадиях присоединяются бульбарные симптомы, прогрессирующая дыхательная недостаточность. Течение более медленное, чем у врожденной формы болезни Верднига-Гоффмана. Пациенты могут доживать до 15-летнего возраста.
Слабость мышц (парез). Тремор.
Диагностика
В диагностическом плане для невролога имеет значение возраст появления первых симптомов и динамика их развития, данные неврологического статуса (в первую очередь наличие двигательных нарушений периферического типа на фоне абсолютно сохранной чувствительности), наличие сопутствующих врожденных аномалий и костных деформаций. Врожденная амиотрофия Верднига-Гоффмана может быть диагностирована неонатологом.
Дифференциальная диагностика
Дифференциальный диагноз проводится с миопатиями, прогрессирующей мышечной дистрофией Дюшенна, боковым амиотрофическим склерозом, сирингомиелией, полиомиелитом, синдромом вялого ребенка, ДЦП, обменными заболеваниями.
С целью подтверждения диагноза проводится электронейромиография — исследование нервно-мышечного аппарата, благодаря которому выявляются характерные изменения, исключающие первично мышечный тип поражения и указывающие на патологию мотонейрона. Биохимический анализ крови не выявляет существенного повышения креатинфосфокиназы, характерного для прогрессирующей мышечной дистрофии. МРТ или КТ позвоночника в редких случаях визуализируют атрофические изменения передних рогов спинного мозга, но позволяют исключить другую спинальную патологию (гематомиелию, миелит, кисту и опухоль спинного мозга).
Окончательный диагноз «амиотрофия Верднига-Гоффмана» устанавливается после получения данных биопсии мышц и генетических исследований. Морфологическое изучение мышечного биоптата выявляет патогномоничную пучковую атрофию мышечных волокон с чередованием зон атрофии миофибрилл и неизмененной мышечной ткани, наличием отдельных гипертрофированных миофибрилл, участков соединительнотканных разрастаний. Проводимый генетиками ДНК-анализ включает прямую и косвенную диагностику. С помощью прямого метода возможна также диагностика гетерозиготного носительства генной аберрации, что имеет важное значение в генетическом консультировании сибсов (братьев и сестер) больных лиц, супружеских пар, планирующих беременность. При этом большую роль играет количественный анализ числа генов локуса СМА.
Дородовый анализ ДНК позволяет снизить вероятность рождения ребенка с болезнью Верднига-Гоффмана. Однако для получения ДНК-материала плода необходимо использовать инвазивные методы пренатальной диагностики: амниоцентез, биопсию хориона, кордоцентез. Амиотрофия Верднига-Гоффмана, диагностированная внутриутробно, выступает показанием к искусственному прерыванию беременности.
Лечение
Этиопатогенетическая терапия не разработана. В настоящее время амиотрофия Верднига-Гоффмана лечится путем улучшения метаболизма периферической нервной системы и мышечной ткани с целью замедлить прогрессирование симптомов. В терапии применяют комбинации препаратов различных фармакологических групп: нейрометаболиты (препараты на основе гидролизата головного мозга свиньи, витамины гр. В, гамма-аминомасляная кислота, пирацетам), облегчающие нервно-мышечную передачу (галантамин, сангвинарин, неостигмин, ипидакрин), улучшающие трофику миофибрилл (глютаминовая к-та, коэнзим Q10, L-карнитин, метионин), улучшающие кровообращение (никотиновая к-та, скополамин). Рекомендована лечебная физкультура и мягкий массаж.
Современное развитие техники позволило несколько облегчить жизнь пациентов и их родственников, благодаря применению автоматизированных инвалидных колясок и портативных аппаратов ИВЛ. Улучшить подвижность пациентов помогают различные методы ортопедической коррекции. Однако основные перспективы в лечении СМА связаны с развитием генетики и поисками возможностей коррекции генетических аберраций методами генной инженерии.
Прогноз
Врожденная амиотрофия Верднига-Гоффмана имеет крайне неблагоприятный прогноз. При ее манифестации в первые дни жизни ребенка, его гибель, как правило, происходит до 6-месячного возраста. При начале клиники после 3-х месяцев жизни, летальный исход наступает в среднем к возрасту 2 года, иногда — к 7-8 годам. Ранняя детская форма характеризуется более замедленным прогрессированием, дети погибают в возрасте 14-15 лет.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Утратил силу — Архив
![]()
РЦРЗ (Республиканский центр развития здравоохранения МЗ РК)
Версия: Архив – Клинические протоколы МЗ РК – 2010 (Приказ №239)
Категории МКБ:
Детская спинальная мышечная атрофия, i тип [Верднига-Гоффмана] (G12.0)
Общая информация
Краткое описание
Спинальная мышечная атрофия Верднига-Гоффманна – нервно-мышечное заболевание, быстропрогрессирующее дегенеративное заболевание клеток передних рогов спинного мозга, наследуемое по аутосомно-рецессивному типу. В основе заболевания лежит неуклонно прогрессирующий дегенеративный процесс в мотонейронах спинного мозга. Признаки заболевания проявляются сразу после рождения или в первые 5 месяцев жизни: быстро прогрессирующая слабость мышц и диффузные, симметричные, преимущественно проксимальные мышечные атрофии, генерализованная гипотония мышц, сухожильная арефлексия, фибриллярные и фасцикулярные подергивания. Проявляются признаками «вялого ребенка».
Протокол “Детская спинальная мышечная атрофия I тип (болезнь Верднига-Гоффманна)”
Коды по МКБ: G 12.0
Мобильное приложение “MedElement”
– Профессиональные медицинские справочники. Стандарты лечения
– Коммуникация с пациентами: онлайн-консультация, отзывы, запись на прием
Скачать приложение для ANDROID / для iOS
Мобильное приложение “MedElement”
– Профессиональные медицинские справочники
– Коммуникация с пациентами: онлайн-консультация, отзывы, запись на прием
Скачать приложение для ANDROID / для iOS
Классификация
1. Злокачественная инфантильная спинальная амиотрофия.
2. Хроническая инфантильная спинальная амиотрофия.
3. Поздне-детская форма спинальной амиотрофии.
Диагностика
Диагностические критерии
Жалобы и анамнез: слабое шевеление плода, с рождения генерализованная мышечная гипотония, атрофии, фасцикулляции мышц, слабый крик, вялое сосание, дисфагии, задержка моторного развития; возможны деформации и контрактуры суставов. В анамнезе – аутосомно-рецессивный тип наследования, дебют заболевания в пренатальный период и первые 5 месяцев жизни. Быстропрогрессирующее течение.
Физикальное обследование: неврологический статус – генерализованная мышечная гипотония, атрофии мышц и фасцикуллярные подергивания в мышцах спины, туловища, проксимальных отделах верхних и нижних конечностей; возможны слабый крик, вялое сосание, дисфагии, фибрилляции мышц языка. Гипорефлексия вплоть до арефлексии; задержка моторного развития. Нарушения чувствительности и психо-речевого развития не характерны.
Лабораторные исследования: общий анализ крови и мочи без патологии.
Инструментальные исследования
ЭМГ мышц: признаки дегенерации периферических мотонейронов (II тип ЭМГ – ритм частокола), уменьшение длительности, амплитуды и изменение формы биопотенциалов, а в тяжелых случаях – полное биоэлектрическое «молчание».
Наличие в биоптатах скелетных мышц групп мелких круглых волокон, гипертрофированных волокон I типа и атрофированных волокон I и II типов.
Показания для консультации специалистов:
1. Психолог.
2. Логопед.
3. Ортопед.
4. Протезист.
5. Врач ЛФК.
6. Физиотерапевт.
Минимум обследования при направлении в стационар:
– общий анализ крови;
– общий анализ мочи;
– АЛТ, АСТ;
– кал на я/г.
Основные диагностические мероприятия:
1. Общий анализ крови (6 параметров).
2. Электромиография.
3. Осмотр психолога, логопеда.
4. Осмотр окулист.
Дополнительные диагностические мероприятия:
1. ЭКГ.
2. УЗИ органов брюшной полости.
3. Компьютерная томография.
Дифференциальный диагноз
Признак | Спинальная мышечная атрофия | Структурные миопатии | ДЦП, атонически-астатическая форма |
Неврологический статус | Генерализованная мышечная гипотония, атрофии мышц и фасцикуллярные подергивания в мышцах спины, туловища, проксимальных отделах верхних и нижних конечностей, гипорефлексия до арефлексии, задержка моторного развития при сохранном психо-речевом развитии | Мышечная гипотония преимущественно в проксимальных отделах конечностей, гипотрофия мышц, гипорефлексия, утомляемость мышц, задержка психо-речевого развития, костно-суставных стоп, пальцев | Мышечная гипотония, гипорефлексия с последующей гиперрефлексией, задержка психо-рече-моторного развития, судороги, деформации (сколиоз, деформации грудной клетки) |
Анамнез | Дебют заболевания в пренатальном периоде и в первые 6 месяцев жизни | Дебют заболевания в неонатальном периоде, задержка моторного развития | Дебют заболевания с рождения |
ЭМГ | Признаки денервации при ЭМГ исследовании, II тип ЭМГ | I тип ЭМГ со снижением амплитуды | I тип ЭМГ со снижением амплитуды |
Течение заболевания | Быстропрогрессирующий характер | Непрогрессирующий или медленно прогрессирующий характер | Постепенная положительная динамика с улучшением двигательных функций |
Тип наследования | Аутосомно-рецессивный | Спорадический и аутосомно-рецессивный | Наследственность не отягощена |
Лечение
Тактика лечения
Цели лечения:
1. Коррекция двигательных нарушений.
2. Обеспечить больному социальную адаптацию.
3. Определить форму спинальной амиотрофии и обеспечить адекватное лечение.
Немедикаментозное лечение:
1. Физиотерапия.
2. Лечебная физкультура.
3. Массаж.
4. Ортопедическая коррекция.
Медикаментозное лечение:
1. Антиоксидатная терапия:
– никотинамид 10-20 мг/сут.;
– аевит 1 кап/сут.
2. Общеукрепляющая терапия: витамины группы В, фолиевая кислота, препараты магния.
3. Аминокислоты: церебролизин, метионин, глутаминовая кислота.
4. Препараты, влияющие на тканевой метаболизм: рибоксин, карнитин, коэнзим Q.
5. Препараты, улучшающие периферическое кровообращение: трентал, теоникол.
6. Стимулирующая терапия: антихолинэстеразные препараты – прозерин, нейромидин, дибазол.
Профилактические мероприятия: полноценное белковое питание с ограничением углеводов, жиров; отдых, профилактика инфекций, контрактур.
Дальнейшее ведение: диспансерный учет у невропатолога по месту жительства. Регулярно занятия ЛФК, проводить курсы поддерживающей терапии через каждые 3-4 месяца.
Основные медикаменты:
1. Аевит, капсулы
2. Актовегин ампулы по 2 мл, 80 мг
3. Винпоцетин (кавинтон), таблетки 5 мг
4. Пиридоксина гидрохлорид, ампулы по 1 мл, 5%
5. Прозерин ампулы по 1 мл, 0,05%
6. Фолиевая кислота, таблетки 0,001
7. Церебролизин, ампулы по 1 мл
8. Цианокобаламин, ампулы по 200 и 500 мкг
Дополнительные медикаменты:
1. Гинко- Билоба, (Танакан) таблетки 40 мг
2. Глицин, таблетки 0,1
3. Дибазол, таблетки 0,02
4. Карнитин хлорид, 20%, 100 мл во флаконах
5. Коэнзим Q
6. Метионин, таблетки 0,25
7. Нейромидин, таблетки 20 мг
8. Нейромультивит, таблетки
9. Неуробекс, таблетки
10. Никотинамид, таблетки 0,025, ампулы 2,5%, 1 мл
11. Пирацетам, ампулы 5 мл, 20%
12. Пирацетам, таблетки 0,2
13. Рибоксин, таблетки 0,2
14. Сермион ампулы и таблетки 5 мг, 10 мг
15. Тиамина хлорид, ампулы 1 мл, 5%
16. Цитохром С, 025%, 4 мл во флаконе, таблетки 10 мг
Индикаторы эффективности лечения:
1. Стабилизация патологического процесса.
2. Повышение мышечного тонуса.
3. Увеличение двигательной активности.
Госпитализация
Показания к госпитализации (плановая): прогрессирующая мышечная слабость, атрофии мышц, дрожание конечностей, ограничение активных движений, мышечная гипотония, отставание в моторном развитии.
Информация
Источники и литература
- Протоколы диагностики и лечения заболеваний МЗ РК (Приказ №239 от 07.04.2010)
- А.С. Петрухин. Неврология детского возраста, Москва 2004
Наследственные болезни нервной системы. Под редакцией Ю.Е. Вельтищева, П.А. Темина, Москва 1998 г.
Е.И. Гусев, Г.С. Бурд, А.С. Никифоров. Неврологические симптомы, синдромы, симптомокомплексы и болезни. Москва 1999
Л.О. Бадалян. Детская неврология. Москва 1998
Джеральд М. Феничел. Педиатрическая неврология. Москва 2004
Л.Р. Зенков, М.А. Ронкин. Функциональная диагностика нервных болезней. Москва 1999
- А.С. Петрухин. Неврология детского возраста, Москва 2004
Информация
Список разработчиков:
№ | Разработчик | Место работы | Должность |
1. | Мухамбетова Гульнара Амерзаевна | Каз.НМУ, кафедра нервных болезней | Ассистент, кандидат медицинских наук |
2. | Кадыржанова Галия Баекеновна | РДКБ «Аксай», психоневрологическое отделение №3 | Заведующая отделением |
3. | Серова Татьяна Константиновна | РДКБ «Аксай», психоневрологическое отделение №1 | Заведующая отделением |
4. | Балбаева Айым Сергазиевна | РДКБ «Аксай», психоневрологическое отделение №3 | Врач-ординатор |
Прикреплённые файлы
Внимание!
Если вы не являетесь медицинским специалистом:
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях “MedElement (МедЭлемент)”, “Lekar Pro”,
“Dariger Pro”, “Заболевания: справочник терапевта”, не может и не должна заменять очную консультацию врача.
Обязательно
обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может
назначить
нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения “MedElement (МедЭлемент)”, “Lekar Pro”,
“Dariger Pro”, “Заболевания: справочник терапевта” являются исключительно информационно-справочными ресурсами.
Информация, размещенная на данном
сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший
в
результате использования данного сайта.
Источник