
Спиноцеребральная дегенерация код по мкб 10

Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Лечение
Названия
Название: Спиноцеребеллярные атаксии.

Спиноцеребеллярные атаксии
Описание
Спиноцеребеллярные атаксии. Группа генетически разнородных наследственных заболеваний неврологического характера, которые проявляются различными расстройствами работы мозжечка и иногда базальных ядер головного мозга. Симптомами этого состояния являются: развитие атаксии и неустойчивой походки, нарушение координации движений и другие неврологические проявления. Диагностика спиноцеребеллярных атаксий производится на основании данных неврологического осмотра, изучения наследственного анамнеза больного, магнитно-резонансной томографии и молекулярно-генетических исследований. Специфического лечения этой патологии на сегодняшний момент не существует, для сохранения оптимального качества жизни больного используют методы поддерживающей и симптоматической терапии.
Дополнительные факты
Спиноцеребеллярные атаксии – группа наследственных неврологических состояний, характеризующихся развитием прогрессирующей дегенерации клеток мозжечка и иногда базальных ядер вплоть до их полной атрофии. Впервые одно из заболеваний этой группы было описано еще в 1891 году немецким невропатологом П. Менцелем, который выявил развитие атаксии, офтальмоплегии и других неврологических нарушений в рамках одной семьи. Дальнейшие исследования показали, что это состояние (известное сейчас как спиноцеребеллярная атаксия 1-го типа) наследуется по аутосомно-доминантному механизму.
В настоящий момент методами современной генетики удалось обнаружить более 20 различных генетических вариантов этого заболевания, при этом более 90% всех случаев обуславливает только 6 из них (1, 2, 3, 6, 7 и 8-й типы). Все формы спиноцеребеллярных атаксий характеризуются аутосомно-доминантным наследованием с явлениями антиципации (усиления выраженности патологии от поколения к поколению) и «отцовской передачи» – более яркой клинической картине заболевания при его наследовании от отца. Поэтому в ряде регионов в общей структуре патологии наблюдается незначительное превалирование больных мужского пола. Общая встречаемость спиноцеребеллярной атаксии колеблется в широких пределах (1-24:100 000), при этом 1-й тип распространен в России и большей части Европы, 2-й – в Индии, 3-й – в Германии и Японии.
Спиноцеребеллярные атаксии
Причины
Несмотря на значительное генетическое и отчасти клиническое разнообразие спиноцеребеллярных атаксий, молекулярные механизмы генетических нарушений при этих заболеваниях очень сходны. Основная причина патологии заключается в изменении количества тринуклеотидных последовательностей (CAG) в кодирующей части ассоциированных с заболеванием генов. Это приводит к увеличению количества аминокислоты глутамина в полученном белке, что изменяет физико-химические свойства протеина и нарушает его функции. В ряде случаев вышеуказанные белки прямо или косвенно участвуют в метаболизме нервной ткани, поэтому изменение их структуры приводит к спиноцеребеллярной атаксии. В настоящее время лучше всего изучены молекулярные механизмы 6 основных разновидностей этого заболевания – данные формы патологии встречаются наиболее часто и в совокупности составляют более 90% случаев спиноцеребеллярной атаксии.
Спиноцеребеллярная атаксия 1. Го типа считается самым распространенным и самым изученным вариантом данной патологии. Ее причиной выступают мутации в гене ATXN1, который располагается на 6-й хромосоме. В норме данный ген имеет не более 36 CAG-повторов, увеличение их количества приводит к развитию заболевания. Продуктом экспрессии гена ATXN1 является особый ДНК-связывающий белок, активно участвующий в метаболизме клеток Пуркинье мозжечка – при наличии мутантной разновидности гена это приводит к появлению агрегантов и постепенной дегенерации, что и становится причиной спиноцеребеллярной атаксии.
Спиноцеребеллярная атаксия 2. Го типа – менее распространенный вариант заболевания, этиология не так тщательно изучена. Причиной патологии является увеличение количества CAG-повторов в гене ATXN2, локализованном на 12-й хромосоме. В здоровом варианте гена количество вышеуказанных последовательностей составляет от 15 до 36, тогда как при спиноцеребеллярной атаксии их может быть свыше 100. Функции белка, который кодируется геном ATXN2, на сегодняшний момент неизвестны.
Спиноцеребеллярная атаксия тип 3 (другое название – болезнь Мачадо-Джозефа в честь двух больных, у которых впервые было описано данное состояние) – причиной этого варианта патологии выступают нарушения в гене ATXN3, расположенном на 14-й хромосоме. В норме количество CAG-повторов в этом гене не превышает 47, при развитии заболевания обнаруживается от 53 до 68 повторов. Данный ген кодирует белок, который предположительно участвует в энергетическом обмене нейронов мозжечка и базальных ядер.
Спиноцеребеллярная атаксия тип 6. Сравнительно редкий вид заболевания, обусловленный дефектами в гене CACNA1A, локализованном на 19 – й хромосоме. Для развития патологии достаточно очень незначительного увеличения количества CAG-повторов – если в нормальном варианте гена их обнаруживают 5-20, то при наличии атаксии – 21-26. Ген CACNA1A кодирует белок-субъединицу кальциевых каналов, расположенных на нейронах мозжечка. Помимо спиноцеребеллярной атаксии, нарушения в гене CACNA1A обуславливают развитие эпизодической атаксии и некоторые наследственные формы мигрени.
Спиноцеребеллярная атаксия тип 7. Данная разновидность патологии вызывается нарушениями структуры гена ATXN7, который располагается на 3 – й хромосоме. У здорового человека количество CAG-повторов составляет не более 35, тогда как при заболевании их количество может достигать нескольких сотен. Функции белка, который кодирует ген ATXN7, на сегодняшний момент изучаются.
Спиноцеребеллярная атаксия тип 8 обусловлена генетическим дефектом гена ATXN8, расположенного на 13-й хромосоме. Как и в других случаях, суть генетического дефекта при этом состоянии заключается в изменении количества тринуклеотидных последовательностей CAG – обычно их около 15-50, тогда как при патологии количество повторов может составлять свыше 1200.
Практически при любом типе спиноцеребеллярной атаксии патологическая форма белка, чрезмерно богатая глутамином, формирует отложения в ядрах или цитоплазме нейронов мозжечка и базальных ядер в виде плотных агрегатов. Этот процесс идет тем быстрее, чем сильнее количество CAG-повторов в ключевом гене отличается от нормы. Этим же объясняется механизм антиципации симптомов спиноцеребеллярной атаксии – в процессе мейоза при образовании половых клеток количество вышеуказанных тринуклеотидных последовательностей может увеличиваться, что приводит к усилению симптомов.
Классификация
Так как подобное явление чаще имеет место при формировании мужских половых клеток, это становится причиной так называемой «отцовской передачи», когда антиципация регистрируется только при передаче заболевания от отца потомству. Многие врачи-генетики полагают, что основная причина спиноцеребеллярных атаксий лежит не в увеличении «гистидиновых» тринуклеотидов, а в делеции так называемых регулирующих триплетов, разделяющих участки CAG-повторов. Например, при первом типе заболевания это CAT, при втором CAA – они регулируют количество CAG-повторов и сохраняют стабильность их количества во время мейоза.
Симптомы
Несмотря на значительное генетическое разнообразие спиноцеребеллярных атаксий, проявления разных типов этого заболевания в целом сходны и различаются только второстепенными деталями – возрастом манифестации, особенностями некоторых симптомов. Практически все формы патологии не регистрируются в детском возрасте – лишь отдельные случаи 1 и 2-го типов были замечены у детей младше 7 лет, средний возраст их манифестации – 18-30 лет. Спиноцеребеллярные атаксии 3, 6 и 7-го типов характеризуются еще более поздним развитием – их манифестация практически всегда происходит у лиц старше 30 лет. Нередко подобные нарушения выявляются и у пожилых людей, что затрудняет дифференциальную диагностику этого состояния с болезнью Паркинсона и другими нейродегенеративными заболеваниями старшего возраста.
Чаще всего развитие спиноцеребеллярной атаксии начинается с появления простой неуклюжести в движениях, особенно при ходьбе, беге. В дальнейшем возникает тремор рук, нарушения походки, паралич глазодвигательных мышц (офтальмоплегия), изменяется почерк больного (становится крупнее, строки неровные). В конечном итоге заболевание приводит к выраженной мозжечковой атаксии, расстройствам пирамидальных и экстрапирамидальных путей, паркинсонизму. Некоторые формы патологии характеризуются выраженными нарушениями зрения – развитием атрофии зрительного нерва, пигментной дегенерации сетчатки и других процессов.
Тремор.
Лечение
Выявление спиноцеребеллярной атаксии производится на основании данных неврологического осмотра, изучения наследственного анамнеза, магнитно-резонансной томографии головного мозга и молекулярно-генетических исследований. При осмотре больных на разных стадиях развития патологии определяются различные по выраженности неврологические нарушения – тремор конечностей, атаксия, изменения речи и голоса, на конечных этапах – дисфагия. Некоторые формы спиноцеребеллярной атаксии сопровождаются достаточно быстрым развитием нарушений зрения, приводящим к полной слепоте. Многолетнее наблюдение за такими больными подтверждает неуклонно прогрессирующее течение заболевания. При изучении наследственного анамнеза могут определяться характерные признаки спиноцеребеллярной атаксии – аутосомно-доминантное наследование, наличие антиципации при передаче болезни от отца.
На МРТ головного мозга при спиноцеребеллярной атаксии обнаруживаются очаги демиелинизации и нейродегенерации в области полушарий, червя мозжечка и базальных ядер. На терминальных стадиях развития заболевания может отмечаться полная атрофия мозжечка. Молекулярно-генетические исследования при спиноцеребеллярной атаксии сводятся к поиску патологически увеличенного количества CAG-повторов в генах, ассоциированных с этим заболеванием. В настоящее время большинство лабораторий мира осуществляет поиск этого дефекта в генах, наиболее часто приводящих к развитию патологии – ATXN1, ATXN2, ATXN3, ATXN7, ATXN8 и CACNA1A.
Специфическое лечение патологии отсутствует, поддерживающая терапия способна несколько замедлить развитие спиноцеребеллярной атаксии, но единого мнения по поводу ее эффективности на сегодняшний момент нет. Применяют витаминотерапию (Е, А, группы В), ноотропные средства, стимуляторы обмена веществ (рибоксин) и метаболизма в нервной ткани. При развитии непроизвольных движений рекомендуют использовать клоназепам и галоперидол. Важную роль в сдерживании прогрессирования спиноцеребеллярной атаксии играет лечебная физкультура – регулярное выполнение правильно подобранного комплекса упражнений позволяет укрепить мышцы и снизить выраженность расстройств равновесия. С этой же целью рекомендуют проведение сеансов лечебного массажа, процедуры электромиостимуляции.
Источник
Рубрика МКБ-10: G11.8
МКБ-10 / G00-G99 КЛАСС VI Болезни нервной системы / G10-G13 Системные атрофии, поражающие преимущественно центральную нервную систему / G11 Наследственная атаксия
Определение и общие сведения[править]
Спиноцеребеллярная атаксия 3 типа
Синонимы: азорская болезнь нервной системы, болезнь Мачадо-Джозефа, офтальмоплегия, болезнь Мачадо, аутосомно-доминантная стрианигральная дегенерация
Спиноцеребеллярная атаксия 3 типа, известная также как болезнь Мачадо-Джозефа, является наиболее распространенным подтипом аутосомно-доминантной мозжечковой атаксии 1-го типа – нейродегенеративное расстройство, характеризующееся атаксией, внешней прогрессирующей офтальмоплегией и другими неврологическими проявлениями.
Распространенность оценивается порядка 1-2 / 100000 со значительными географической и этнической вариативностью: самый высокий показатель распространенности был обнаружен на Азорских островах (1/239), промежуточные показатели распространенности отмечены в Португалии, Германии, Нидерландах, Китае и Японии. Самая низкая распространенность в Северной Америке, Австралии и Индии. Точные оценки распространенности не доступны, тем не менее, в наиболее генетически характеризуемых популяциях, спинномозжечковая атаксия 3 типа составляет до 72% семей с атаксией. Около 600 случаев заболевания было опубликовано в литературе.
Этиология и патогенез[править]
Заболевание связано с мутацией CAG-повторов с феноменом антиципации гена ATXN3 (14q21). Нормальная длина повтора 13-41, тогда как при данной патолгии их больше 56.
Клинические проявления[править]
Спиноцеребеллярная атаксия 3 типа делится на 3 формы.
Тип 1 проявляется атаксией, офтальмоплегией, пирамидальными расстройствами (спастичность и гиперрефлексия) и экстрапирамидными расстройствами, включая дистонию и другие двигательные нарушения. Манифестирует в подростковом возрасте.
Тип 2 (тип Тома) обнаруживается в зрелом возрасте, проявляется атаксией, спастичностью и дистонией. Составляет 57% случаев болезни Мачадо.
3-й тип манифестирует в возрасте после 40 лет и включает в себя симптомы офтальмоплегии и дефекта клеток переднего рога – фасцикуляции, атрофия и слабость.
Паркинсонизм может также быть признаком спиноцеребеллярной атаксии 3 типа. Часто упускается из виду, но общим признаком заболевания является нарушение температурной чувствительности всего тела.
Другая наследственная атаксия: Диагностика[править]
Диагноз основывается на клинической картине, семейном анамнезе и в конечном счете на данных генетического тестирования.
Пренатальная диагностика проводится у пациентов с семейной историей патологии.
Дифференциальный диагноз[править]
Дифференциальнай диагноз очень широк и включает в себя другие виды спинномозжечковых атаксий.
Другая наследственная атаксия: Лечение[править]
Лечение симтоматическое. Паркинсонизм, синдром усталых ног, спастичность, нарушения сна и депрессия – поддаются фармакологическому контролю. Дистония и спастичность может корректироваться локальными инъекциями ботулинического токсина. Профессиональная, логопедическая и физическая терапии имеют важное значение.
Прогноз
Прогноз неблагоприятный, но известны случаи, когда пациенты прожили несколько десятилетий после появления симптомов.
Профилактика[править]
Прочее[править]
Аутосомно-доминантная мозжечковая атаксия
Аутосомно-доминантная мозжечковая атаксия описывает клинически и генетически гетерогенную группу нейродегенеративных заболеваний, характеризующихся медленно прогрессирующей атаксией походки, позы и конечностей, дизартрией и/или глазодвигательными расстройствами.
Аутосомно-доминантная мозжечковая атаксия вызывается дегенерацией мозжечка при отсутствии сопутствующих заболеваний. Дегенеративный процесс может быть ограничен мозжечком при аутосомно-доминантной мозжечковой атаксии 3-го типа; может дополнительно включать в себя нарушения сетчатки (тип 2); зрительный нерв, мосто-мозжечковую систему, базальные ганглии, кору головного мозга, спинной мозг или периферические нервы – при аутосомно-доминантной мозжечковой атаксии 1-го типа. При 4-м типе патологии мозжечковый синдром сопровождается с эпилепсией.
Распространенность всех типов аутосомно-доминантной мозжечковой атаксии оценивается в 1/37000.
Наследственная эпизодическая атаксия
Наследственная эпизодическая атаксия представляет собой группу неврологических расстройств, характеризующихся повторяющимися эпизодами атаксии и головокружений, которые могут со временем прогрессировать. Слабость, дистония и атаксия иногда присутствуют в межприступном периоде. Семь типов эпизодической атаксии были описаны на сегодняшний день (ЭА1 : ЭА7), но большинство зарегистрированных случаев относится наследственной эпизодической атаксии 1-го или 2-го типов.
Распространенность оценивается 1-9 / 100 000. Наследование аутосомно-доминантное.
Эпизодическая атаксия тип 4
Синонимы: периодическая вестибулоцеребеллярная атаксия, вестибуло-мозжечковый синдром
Эпизодическая атаксия 4-го типа является очень редкой формой наследственной эпизодической атаксии, характеризуется эпизодической атаксией с поздним началом, повторяющимися приступами головокружения и диплопии.
Источники (ссылки)[править]
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
- Прогноз
Названия
Название: Оливопонтоцеребеллярные дегенерации.
Оливопонтоцеребеллярные дегенерации
Описание
Оливопонтоцеребеллярные дегенерации. Наследственные дегенеративные заболевания ЦНС, объединенные сходной локализацией патологического процесса в мозжечке, нижних оливах и мосте головного мозга. Клиника складывается из мозжечкового синдрома, экстрапирамидных расстройств, когнитивных и психических нарушений. Диагностируются оливопонтоцеребеллярные дегенерации на основании анамнеза, генеалогического исследования, данных неврологического и психологического обследования, результатов КТ и МРТ головного мозга. Терапия симптоматическая, включает нейропротекторные и общеукрепляющие фармпрепараты, ЛФК, массаж. Прогноз неблагоприятный.
Дополнительные факты
Прогрессирующий мозжечковый синдром в сочетании с экстрапирамидными нарушениями и психическими расстройствами был описан Дежерином и Томасом в 1900 году. В последующем были выделены несколько форм данной патологии, объединенные в единую группу заболеваний, получившую название «оливопонтоцеребеллярные дегенерации» (ОПЦД). По сути, речь идет о заболеваниях с единой локализацией мультифокальной дегенерации и гибели нейронов. Именно преимущественное расположение дегенеративных процессов легло в основу названия этой группы поражений ЦНС (с латинского oliva — олива, pont(is) — мост, cerebellum — мозжечок). В ряде случаев наблюдается поражение каудальных черепно-мозговых нервов (IX, X, XI, XII пар), реже — передних рогов спинного мозга и проводящих трактов.
Оливопонтоцеребеллярные дегенерации входят в группу дегенеративных поражений ЦНС, к которой относятся болезнь Паркинсона, рассеянный склероз, болезнь Альцгеймера, лейкодистрофии, болезнь Пика, спинальные амиотрофии и мн. Тд Отмечается аутосомное рецессивное и доминантное наследование, спорадические случаи. Возраст манифестации клинических проявлений варьирует в пределах от 11 до 80 лет, наиболее часто дебют происходит в четверной или пятой декаде жизни.
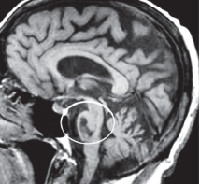
Оливопонтоцеребеллярные дегенерации
Причины
Точные представления о этиопатогенезе ОПЦД пока не сформированы. Поиск генетического субстрата дегенераций привел к выявлению нарушений в локусе 6p22-23 (при дегенерации Менделя) и в локусе 12q23-24 (при дегенерации Фиклера-Винклера) в виде увеличения числа тринуклеотидных повторов. У ряда пациентов наблюдается недостаточность дегидрогеназы глутаминовой кислоты, необходимой для метаболизма глутамата. Последний в качестве медиатора активирует передачу возбуждения от мозжечковой коры к клеткам Пуркинье, аксоны которых формируют эфферентные (нисходящие) мозжечковые тракты. Избыточное накопление глутамата при дефиците дегидрогеназы оказывает нейротоксический эффект, который, возможно, является основной причиной дегенеративных изменений клеток Пуркинье.
Основными морфологическими признаками ОПЦД выступают: асимметричные атрофические изменения белого вещества мозжечковых полушарий и в меньшей степени его червя, дегенерация средней и нижней мозжечковых ножек, глиоз и сморщивание ядер моста и олив. Атрофия коры мозжечка с утратой клеток Пуркинье отмечается на более поздних стадиях ОПЦД. Типична полная интактность верхней ножки, узелка червя (nodulus) и клочка (flocculus) мозжечка. Гистологический анализ пораженных церебральных тканей выявляет дистрофически-дегенеративные изменения нейронов, разрастание глии, демиелинизацию нервных волокон.
Классификация
В настоящее время в клинической неврологии известны 5 основных типов оливопонтоцеребеллярных дегенераций. Отдельно выделяют синдром Шая-Дрейджера, который наряду с оливопонтоцеребеллярной дегенерацией включает диффузную церебральную атрофию и дегенерацию стрионигральных структур.
Tип I Менделя. Аутосомно – доминантная ОПЦД с дебютом после 11 лет и до 60 – летнего возраста. Характерна мозжечковая атаксия, гиперкинезы, гипотония мышц, дисфагия. Реже встречаются пирамидные расстройства (парезы конечностей, гипестезия).
Tип II Фиклера. Винклера – аутосомно – рецессивная ОПЦД, манифестирующая с третьей декады жизни до 80 – летнего возраста. Протекает без нарушений чувствительности и парезов. Глубокие рефлексы сохранены.
Tип III ОПЦД с ретинальной дегенерацией. Аутосомно – доминантная форма, поражающая преимущественно лиц молодого возраста. Характеризуется мозжечковым синдромом и гиперкинезами в сочетании с прогрессирующим падением зрения вследствие пигментной ретинопатии.
Tип IV Шута. Хайкмана – аутосомно – доминантная ОПЦД детского и молодого возраста. Типичные для всех ОПЦД мозжечковые расстройства сочетаются с поражением каудальных черепных нервов (бульбарные симптомы) и задних столбов спинного мозга (нарушение глубокой чувствительности).
Tип V ОПЦД с деменцией, экстрапирамидными знаками и офтальмоплегией. Наследуется аутосомно-доминантно. Представляет собой комбинацию указанных в названии синдромов и мозжечковой атаксии.
Симптомы
Базисным клиническим проявлением ОПЦД является мозжечковая атаксия. Дебют заболевания знаменуется появлением легкой неустойчивости, неуклюжести при беге и быстрой ходьбе. Прогрессирование этих симптомов приводит к выраженным расстройствам походки и статики. Ходьба затруднена, сопровождается падениями, во избежание которых пациенты широко расставляют ноги во время ходьбы. Грубые нарушения равновесия обуславливают колебания туловища пациента, когда он стоит или сидит. Позднее присоединяется дискоординация в конечностях: адиадохокинез, гипер- и дисметрия, крупноразмашистый почерк. Атаксия в конечностях сопровождается дрожанием головы и интенционным тремором. Наблюдается горизонтальный нистагм. Одновременно с атаксией в конечностях появляется типичная мозжечковая дизартрия, т. Н. «скандированная речь». Как правило, происходит повышение рефлексов, в отдельных случаях — их понижение.
Возможны расстройства глотания (дисфагия), гиперкинезы, симптомы вторичного паркинсонизма, лицевой парез, пирамидная недостаточность. При поражении ядер каудальных черепных нервов возникает офтальмоплегия, бульбарный паралич. В большинстве случаев оливопонтоцеребеллярные дегенерации протекают с недержанием мочи. В эмоциональной сфере преобладает снижение: вялость, безынициативность, тупость. Обычно отмечаются значительные когнитивные нарушения и деменция. Характерны психические расстройства: галлюцинаторный синдром, депрессия, фобические расстройства, эпизоды психомоторного возбуждения, спутанность сознания. Иногда их появление предшествует развитию мозжечковой атаксии.
Вялость. Галлюцинации. Тремор. Тремор головы.
Диагностика
Постановка диагноза требует сопоставления времени и симптомов дебюта заболевания, данных неврологического статуса (сочетание мозжечковых нарушений с гиперкинезами) и нейропсихологического обследования (наличие когнитивного снижения, отклонения в эмоциональной сфере) с результатами нейровизуализации.
Дифференциальная диагностика
Дифференцировать оливопонтоцеребеллярные дегенерации необходимо от атаксии Пьера-Мари и атаксии Фридрейха, опухолей мозжечка, прогрессирующих вариантов рассеянного склероза, дисметаболических заболеваний с мозжечковым синдромом (например, болезни Рефсума).
Компьютерная томография малоинформативна, поскольку определяет преимущественно неспецифические изменения церебральных структур: расширение желудочков и субарахноидальных пространств. Специфичным признаком, регистрируемым при помощи КТ головного мозга, является уменьшение толщины передней мозжечковой ножки. Более полно диагностировать оливопонтоцеребеллярные дегенерации позволяет МРТ головного мозга. С ее помощью можно визуализировать атрофические изменения в мосте и продолговатом мозге. Для определения типа наследования патологии необходима консультация генетика и генеалогическое исследование. При подозрении на ОПЦД I или II типов возможна ДНК-диагностика. Пациенты с нарушением зрения нуждаются в консультации офтальмолога.
Лечение
Специфическая терапия ОПЦД пока не найдена, поэтому неврологами осуществляется симптоматическое лечение, т. Е. Направленное на уменьшение конкретных клинических проявлений. Показаны нейрометаболиты и общеукрепляющие средства: витамины гр. В, витамин С и тд Для нормализации мышечного тонуса проводится массаж и ЛФК. С целью уменьшения выраженности нарушений координации рекомендованы специальные упражнения для ее тренировки. При синдроме паркинсонизма показаны центральные холинолитики: диэтазина гидрохлорид, тригексифенидил.
Прогноз
Несмотря на проводимое лечение, оливопонтоцеребеллярные дегенерации имеют неуклонно прогрессирующее течение. Длительность заболевания в среднем колеблется в пределах 10-15 лет, иногда достигает 20 лет. Причиной летального исхода, как правило, являются интеркуррентные инфекции (застойная пневмония, сепсис).
Источник