
Тип наследования синдрома гарднера диффузный полипоз кишечника

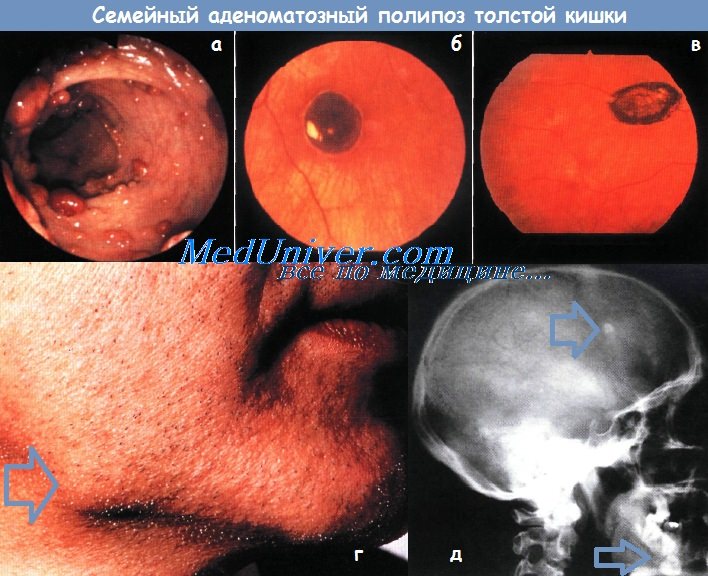
Диффузный полипоз кишечника как наследственный синдром и предраковое заболеваниеПолипозные синдромы характеризуются наличием/развитием множественных полипов в различных отделах желудочно-кишечного тракта, но часто сопровождаются другими проявлениями. Некоторые полипозные синдромы неизбежно приводят к злокачественной трансформации полипов и развитию рака (например, САТК, АСАТК); другие – не связаны с развитием рака напрямую, но могут служить индикаторами повышенного риска возникновения некоторых кишечных или внекишечных опухолей. Семейный аденоматоз толстой кишки (САТК, АСАТК)• Фенотип: множественные аденоматозные полипы по всей толстой кишке, периампулярные дуоденальные полипы, полипы желудка, внекишечные проявления (десмоиды и т.д.). • Тип наследования: аутосомно-доминантный, почти полная пенетрантность гена. • Локализация гена: ген аденоматозного полипоза толстой кишки (АРС) локализован в хромосоме 5q21. • Обычное течение заболевания: почти 100% развитие рака толстой кишки в возрасте 40-50 лет, в 3-12% случаев – периампулярный рак. • Ассоциированные опухоли: рак толстой кишки, рак в резервуаре, периампулярная аденокарцинома, десмоидные опухоли, рак щитовидной железы. • Варианты:
MYH-ассоциированный полипоз (МАП)• Фенотип: часто не отличим от САТК, за исключением несколько меньшего числа полипов толстой кишки, внекишечные проявления присутствуют, но менее выражены, чем при САТК: полипы верхних отделов ЖКТ (=> периампулярный рак), остеомы, изменения зубов, наследственная гипертрофия пигментного эпителия сетчатки и др. • Тип наследования: аутосомно-рецессивный, почти полная пенетрантность гена. • Локализация гена: ген репарации MYH, хромосома 1р34-32. • Обычное течение заболевания: диагноз МАП устанавливается в возрасте около 50 лет, почти в 100% случаев к 65 годам развивается рак. • Ассоциированные опухоли: рак толстой кишки, периампулярная аденокарцинома, рак молочной железы, рак щитовидной железы. • Консультация: оба родителя и все дети являются носителями гена. Синдром Пейтца-Егерса• Фенотип: гамартомные полипы ЖКТ, в частности его верхних отделов, отложение меланина в коже (например, около рта, на слизистой щек и т.д.).
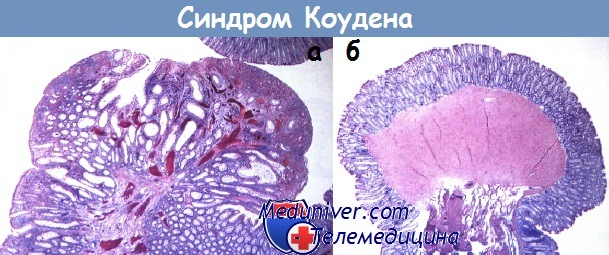
Ювенильный полипоз• Фенотип: гамартомные полипы, в 15% сочетающиеся с врожденными дефектами развития. Синдром Коудена• Фенотип: синдром множественных гамартом из эктодермальных и в меньшей степени из эндодермальных элементов (трихолеммома — 80% случаев, макроцефалия – 40% случаев, полипоз ЖКТ – только 35% случаев, доброкачественные заболевания щитовидной и молочной желез).
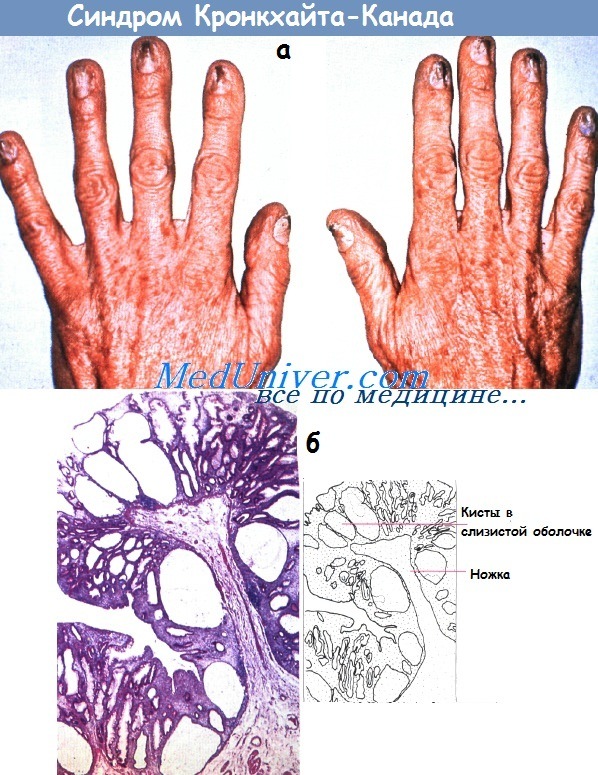
Синдром Баннаяна-Райли-Рувалькаба (ранее синдром Рувалькаба-Майра-Смита)• Фенотип: прогрессивный рост до/после рождения, макроцефалия, задержка умственного и психомоторного развития, другие аномалии; множественные гамартомные полипы ЖКТ; липомы; пигментные пятна на гениталиях. Синдром Кронкхайта-Канада• Фенотип: диффузный полипоз всего ЖКТ (за исключением пищевода), эктодермальные аномалии (например, алопеция, ониходистрофия, гиперпигментация кожи).
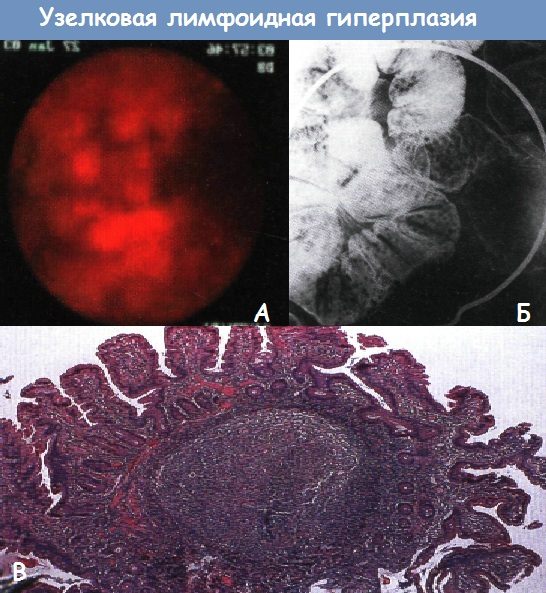
Синдром гиперпластического полипоза• Фенотип: множественные гиперпластические полипы всех отделов толстой и прямой кишки, включая крупные (> 1 см) полипы, локализованные прокси-мальнее сигмовидной кишки. Нодулярная лимфоидная гиперплазия• Фенотип: множественные лимфоидные полипы всех отделов толстой и прямой кишки. Дополнительные исследования при полипозах кишечникаГенетическая консультация и тестирование => оценка индивидуального и семейного риска.
– Также рекомендуем “Семейный аденоматоз толстой кишки (САТК) – причины, признаки, лечение” Оглавление темы “Опухоли толстой кишки”:
|

Источник
Синдром Гарднера – это наследственное заболевание, сопровождающееся полипозом толстого кишечника в сочетании с доброкачественными неоплазиями кожи, костей и мягких тканей. Может долгое время протекать бессимптомно. Возможны вздутие живота, урчание и расстройства стула. В некоторых случаях полипоз кишечника осложняется кровотечением или кишечной непроходимостью. Отмечается высокая вероятность развития колоректального рака. Заболевание диагностируется на основании жалоб, семейного анамнеза, данных осмотра, рентгенографии, КТ, МРТ, УЗИ, эндоскопии и других исследований. Лечение – эндоскопическая полипэктомия или резекция пораженных отделов кишечника.
Общие сведения
Синдром Гарднера – редкая генетически обусловленная патология, при которой наблюдается диффузный полипоз толстого кишечника в сочетании с доброкачественными опухолями костей и мягких тканей (остеомами, фибромами, нейрофибромами, эпителиальными кистами и другими неоплазиями). Полипозом при синдроме Гарднера преимущественно поражаются прямая и сигмовидная кишка, однако полипы могут выявляться в других отделах кишечника. Впервые был описан американским врачом и генетиком Е. Дж. Гарднером в 1951 году. С тех пор в специальной литературе появились упоминания более чем о ста случаях данного заболевания. Риск малигнизации полипов толстой кишки с развитием колоректального рака в течение жизни составляет около 95%. Лечение проводят специалисты в сфере клинической проктологии, гастроэнтерологии, онкологии, ортопедии, стоматологии и челюстно-лицевой хирургии.

Синдром Гарднера
Причины
Синдром Гарднера передается по аутосомно-доминантному типу. Выраженность кишечных и внекишечных клинических проявлений может сильно варьировать. Первые симптомы синдрома Гарднера обычно появляются у детей старше 10 лет. Возможно позднее начало с образованием первых опухолей в возрасте старше 20 лет. В отдельных случаях наряду с полипозом толстого кишечника, остеомами и мягкотканными новообразованиями у больных синдромом Гарднера обнаруживаются полипы тонкого кишечника, желудка и двенадцатиперстной кишки.
Симптомы
Синдром Гарднера включает в себя характерную триаду: диффузный полипоз нижних отделов толстого кишечника, остеомы плоских и трубчатых костей, различные доброкачественные опухоли кожи и мягких тканей. При умеренном количестве и небольшом размере полипов кишечные проявления синдрома Гарднера могут отсутствовать или быть слабо выраженными. В подростковом или юношеском возрасте больные обычно впервые обращаются к врачам в связи с появлением доброкачественных костных и мягкотканных опухолей.
Остеомы при синдроме Гарднера могут локализоваться как в плоских, так и в трубчатых костях. Часто наблюдается поражение костей лицевого черепа, сопровождающееся обезображиванием. Возможно смещение и даже выпадение зубов. Через некоторое время после появления рост остеом у больных синдромом Гарднера прекращается, опухоли не озлокачествляются. Неоплазии мягких тканей отличаются большим разнообразием. Особенно часто выявляются липомы, дерматофибромы, нейрофибромы и эпителиальные кисты. Реже встречаются атеромы, лейомиомы и другие новообразования. Мягкотканные опухоли при синдроме Гарднера также протекают доброкачественно, малигнизация отсутствует.
Полипы толстой кишки при синдроме Гарднера нередко становятся случайной находкой при проведении исследований ЖКТ по другим поводам либо обнаруживаются в процессе расширенного обследования, назначенного в связи с появлением множественных мягкотканных и костных неоплазий. В течении синдрома Гарднера можно выделить три стадии поражения кишечника. На первой стадии заболевание протекает бессимптомно. На второй пациенты отмечают дискомфорт в животе, вздутие, урчание и периодические нарушения стула. В каловых массах могут обнаруживаться примеси крови и слизи.
На третьей стадии у больных синдромом Гарднера выявляются выраженный болевой синдром, постоянный метеоризм, обильные примеси слизи и крови в испражнениях, снижение веса, повышенная утомляемость, эмоциональная лабильность, нарушения электролитного и белкового обмена. У многих пациентов с синдромом Гарднера развивается анемия, обусловленная небольшими по объему, но часто повторяющимися кровотечениями из нижних отделов ЖКТ. В отдельных случаях у больных развиваются неотложные состояния, требующие экстренной медицинской помощи – обильные кишечные кровотечения или кишечная непроходимость.
Диагностика
Диагноз устанавливается на основании семейного анамнеза (наличия синдрома Гарднера у близких родственников), клинической картины, включающей в себя характерную триаду, и данных дополнительных исследований. При проведении физикального осмотра врач отмечает наличие множественных костных и мягкотканных опухолей различной локализации. У некоторых больных синдромом Гарднера выявляются деформации лица, обусловленные остеомами лицевого черепа. При пальпации костей туловища и конечностей могут обнаруживаться опухолевидные образования костной плотности. При поражениях легкой степени количество неоплазий может быть незначительным, что затрудняет диагностику.
При пальпации живота наблюдается болезненность в левой подвздошной области. На первой стадии поражения кишечника данный симптом может отсутствовать. При проведении пальцевого ректального исследования на слизистой прямой кишки больных синдромом Гарднера обнаруживаются множественные узлы. На контрастных рентгеновских снимках такие узлы отображаются в виде дефектов наполнения. При узлах небольшого размера (менее 1 см) информативность контрастного рентгенологического исследования снижается. В ходе ректороманоскопии выявляются полипы в прямой и ободочной кишке. Количество полипов может сильно варьировать.
У некоторых пациентов с синдромом Гарднера отмечаются ограниченные поражения отдельных участков кишки. В отличие от рентгенографии, эндоскопическое исследование дает возможность диагностировать полипы любого размера, в том числе – мелкие (диаметром от 1-2 мм). Для уточнения характера и распространенности костных опухолей при синдроме Гарднера осуществляют рентгенографию. При мягкотканных новообразованиях назначают КТ, МРТ или УЗИ области поражения. При необходимости выполняют биопсию полипов, остеом и мягкотканных новообразований.
Дифференциальную диагностику синдрома Гарднера врачи-проктологи проводят с обычными множественными полипами и другими формами семейного полипоза. Для разных вариантов наследственного полипоза характерны определенные отличия в преимущественной локализации полипов (поражение всего толстого кишечника, поражение дистальных отделов толстой кишки), характере патологических изменений костей и мягких тканей. Для уточнения этих различий перед постановкой окончательного диагноза проводят детальный внешний осмотр, осуществляют ирригоскопию и колоноскопию.
Лечение синдрома Гарднера
Лечение только хирургическое. Поскольку риск озлокачествления костных и мягкотканных неоплазий отсутствует, решение о проведении оперативных вмешательств принимают при наличии косметического или функционального дефекта. Полипоз толстого кишечника при синдроме Гарднера рассматривается, как облигатный предрак, поэтому многие врачи считают целесообразным проведение операции до появления признаков малигнизации. При небольшом количестве полипов возможна эндоскопическая полипэктомия.
При синдроме Гарднера с выраженным диффузным полипозом показана резекция пораженного участка кишечника или тотальная колэктомия с наложением илеостомы либо формированием илеоректального анастомоза (при отсутствии полипов прямой кишки). Хирургическое вмешательство рекомендуют проводить в возрасте 20-25 лет. Из-за калечащего характера операции молодые пациенты с синдромом Гарднера нередко отказываются от данного вмешательства. В подобных случаях показано динамическое наблюдение с проведением колоноскопии через каждые 6-8 месяцев.
Некоторые врачи являются сторонниками выжидательной тактики и считают, что колэктомию при синдроме Гарднера следует проводить только при появлении признаков озлокачествления или при часто повторяющихся кровотечениях с развитием анемии. Показанием к экстренному оперативному вмешательству при синдроме Гарднера являются обильное кишечное кровотечение и кишечная непроходимость.
Прогноз и профилактика
При своевременном адекватном лечении прогноз при синдроме Гарднера достаточно благоприятный. Тяжесть течения определяется выраженностью полипоза и локализацией внекишечных опухолей. Родителям, имеющим родственников, страдающих данным заболеванием, в период планирования беременности рекомендуют обратиться за медико-генетической консультацией.
Источник