
У людини з каріотипом xxy синдром

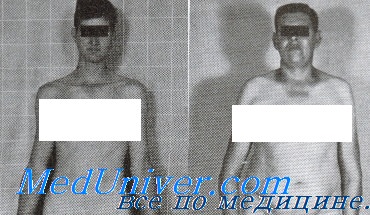
Синдром Кляйнфелтера (47,XXY): признаки, фенотипФенотип синдрома Кляйнфелтера — первой описанной аномалии половых хромосом у человека — показан на рисунке. Пациенты — высокие и худощавые, имеют сравнительно длинные ноги. Они нормально развиваются физически до половой зрелости, когда становятся очевидными признаки гипогонадизма. Половая зрелость наступает в нормальном возрасте, но яички остаются небольшими, а вторичные половые признаки недоразвитыми. Гинекомастия характерна для некоторых пациентов; вследствие этого риск рака груди в 20-50 раз выше, чем у мужчин с обычным кариотипом 46,XY. Пациенты с синдромом Кляйнфелтера почти всегда бесплодны из-за нарушений развития половых клеток, и пациенты часто клинически диагностируют при обследовании по поводу бесплодия. Синдром Кляйнфелтера сравнительно часто встречают среди бесплодных мужчин (около 3%) и мужчин с олигозооспермией и азооспермией (5-10%). В зрелости устойчивый дефицит андрогенов может приводить к уменьшению мышечной массы, снижению либидо и остеопорозу.
Встречаемость в популяции — по крайней мере, 1 на 1000 новорожденных мальчиков (1 на 2000 родов в целом). Как указано ранее, одна из двух Х-хромосом инактивируется. Из-за сравнительно мягкого и вариабельного фенотипа предполагают, что многие случаи остаются не выявленными. Около половины случаев синдрома Кляйнфелтера вызвано ошибкой в первом делении отцовского мейоза — неудачной рекомбинацией Xp/Yp в псевдоаутосомной области. Среди случаев материнского происхождения большинство — результат нарушений в первом и, реже, во втором делении мейоза или ошибок в постзиготических митозах, ведущих к мозаицизму. В случаях, связанных с первым делением материнского мейоза, отмечают связь с возрастом матери. Примерно 15% пациентов с синдромом Кляйнфелтера имеют мозаичные кариотипы. Мозаичные пациенты имеют вариабельные фенотипы, у некоторых могут быть нормально сформированные яички. Наиболее частый мозаичный кариотип — 46,XY/47,XXY, вероятно в результате утраты одной Х-хромосомы в эмбрионах XXY в ранних постзиготических делениях.
Есть несколько вариантов синдрома Кляйнфелтера, с иными кариотипами, нежели 47,XXY, это 48,XXYY, 48.XXXY и 49.XXXXY. Как правило, дополнительные Х-хромосомы (даже если они по большей части неактивны) вызывают соотвествен-но большие изменения в фенотипе, с большей степенью дисморфичности, более выраженными нарушениями полового развития и более серьезной умственной отсталостью. Хотя среди пациентов с этими и другими половыми хромосомными анеуплоидиями существует широкое фенотипическое разнообразие, отмечены некоторые важные фенотипические различия между пациентами с синдромом Кляйнфелтера и хромосомно нормальными мужчинами. У мужчин с кариотипом 46.XXY снижены вербальные способности и понимание, а также результаты интеллектуальных тестов (IQ). Пациенты с синдромом Кляйнфелтера имеют многократно повышенный риск развития затруднений в обучении, особенно в чтении, которые могут потребовать специальной педагогической коррекции. Синдром Кляйнфелтера очень часто бывает среди мальчиков, требующих специального образования. Многие больные мальчики имеют сравнительно низкие психосоциальные установки, частично связанные с восприятием своего тела. Языковые трудности могут вести к робости, неуверенности и психологической незрелости. – Вернуться в содержание раздела “генетика” на нашем сайте Оглавление темы “Хромосомные патологии”:
|

Источник
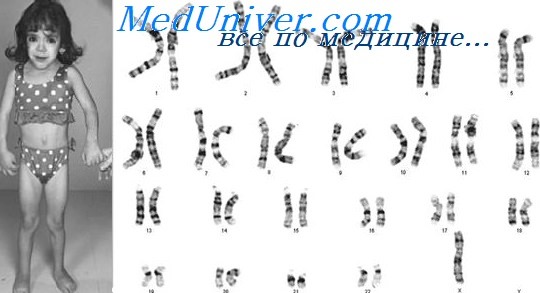
Синдром 48, XXYY — это аномалия хромосом, при которой у человека имеется дополнительная X и Y-хромосома. Клетки человека обычно содержат две половые хромосомы, одну от матери и одну от отца. Обычно женщины имеют две Х-хромосомы (XX), а мужчины имеют одну Х и одну Y-хромосому (XY). Появление по крайней мере одной Y-хромосомы с правильно функционирующим геном SRY делает человека физиологически мужчиной. Следовательно, люди с XXYY являются генотипически мужчинами. Мужчины с синдромом XXYY имеют 48 хромосом вместо типичных 46. По оценкам, он случается в 1 случае на каждые 18 000–40 000 новорожденных[2].
Симптомы[править | править код]
Симптомы этого синдрома включают в себя:
Причины[править | править код]
Обычно люди имеют 46 хромосом в каждой клетке. Две из 46 хромосом, известные как X и Y, называются половыми хромосомами, потому что они помогают определить, будут ли у человека развиваться мужские или женские половые признаки. Женщины обычно имеют две Х-хромосомы (46, XX), а мужчины имеют одну Х-хромосому и одну Y-хромосому (46, XY). Синдром 48, XXYY возникает из-за наличия дополнительной копии обеих половых хромосом в каждой из клеток. Дополнительные копии генов на Х-хромосоме препятствуют половому развитию мужчины, препятствуя нормальному функционированию яичек и снижая уровень тестостерона. Многие гены обнаруживаются только в X или Y хромосоме, но гены в областях, известных как псевдоавтосомные области, присутствуют в обеих половых хромосомах. Дополнительные копии генов из псевдоаутосомных областей лишних X и Y хромосом вносят вклад в симптомы синдрома; однако конкретные гены не были идентифицированы[3][4].
Генетика[править | править код]
Синдром не наследуется; Он обычно возникает случайно при формировании репродуктивных клеток (яйцеклетки и сперма). Ошибка в клеточном делении, называемая недисключением, приводит к появлению репродуктивной клетки с аномальным количеством хромосом. При синдроме 48, XXYY, дополнительные половые хромосомы почти всегда происходят из сперматозоидов. Несоответствие может привести к тому, что сперматозоид приобретет две дополнительные половые хромосомы, в результате чего сперматозоид будет иметь три половые хромосомы (одну Х и две Y-хромосомы). Если этот сперматозоид оплодотворяет нормальную яйцеклетку одной Х-хромосомой, у получающегося ребенка будет две Х-хромосомы и две Y-хромосомы в каждой из клеток его тела.
В небольшом проценте случаев синдром 48, XXYY является следствием непереключения половых хромосом у эмбриона 46, XY очень скоро после оплодотворения. Это означает, что нормальный сперматозоид с одной Y-хромосомой оплодотворил нормальную яйцеклетку с одной X-хромосомой, но сразу после оплодотворения, несопряжение половых хромосом заставило эмбрион получить две дополнительные половые хромосомы, в результате чего зародыш имеет кариотип 48, XXYY[3].
Диагностика[править | править код]
Для диагностики проверяют кариотип[5].
Лечение[править | править код]
Пациенты обычно должны наблюдаться у эндокринолога. При наличии гипогонадизма лечение тестостероном следует рассматривать у всех людей, независимо от когнитивных способностей, из-за положительного влияния на здоровье костей, мышечный тонус, усталость и выносливость, а также с возможными преимуществами для психического здоровья / поведения[2].
У большинства детей с XXYY наблюдаются некоторые задержки в развитии и трудности в обучении. Следовательно, эти аспекты должны наблюдаться: психология (когнитивное и социально-эмоциональное развитие), логопедия, трудотерапия и физиотерапия. Следует организовать консультации с развивающими педиатрами, психиатрами или неврологами для разработки плана лечения, включающего терапию, поведенческие вмешательства, образовательную поддержку и психотропные препараты для поведенческих и психиатрических симптомов. Общие диагнозы, такие как неспособность к обучению, СДВГ, расстройства аутистического спектра, перепады настроения, тиковые расстройства и другие проблемы психического здоровья, должны быть рассмотрены, обследованы и пролечены. В этой группе наблюдаются хорошие реакции на стандартные медикаментозные методы лечения от невнимательности, импульсивности, тревоги и нестабильности настроения, и такое лечение может положительно влиять на успеваемость, эмоциональное благополучие в долгосрочной перспективе. Плохая координация мелкой моторики может сделать письмо медленным и трудоемким занятием, и трудотерапия и клавиатура должны быть введены в раннем возрасте, чтобы облегчить школьную работу и навыки самопомощи. Образовательные трудности должны оцениваться с помощью полной психологической оценки, чтобы выявить расхождения между устными и служебными навыками и выявить индивидуальные академические потребности. Языковые навыки часто страдают в течение всей жизни, и в зрелом возрасте могут потребоваться вмешательства в области логопедической терапии, направленные на развитие выразительных языковых навыков, диспраксии и др. Адаптивные навыки представляют собой затруднения, требующие поддержки на уровне сообщества почти для всех людей в зрелом возрасте[2]. Могут потребоваться дополнительные рекомендации по лечению, основанные на индивидуальных сильных и слабых сторонах синдрома XXYY[6].
Прогноз[править | править код]
Пациенты имеют практически нормальную продолжительность жизни, но им требуются регулярное медицинское наблюдение[3][7].
История[править | править код]
Первое опубликованное сообщение о мальчике с кариотипом 48, XXYY было опубликовано Сильфестом Малдалом и Чарльзом Оки в Манчестере, в 1960 году[8]. Он был описан у 15-летнего мальчика с психическими расстройствами, у которого были признаки синдрома Клайнфелтера; однако, тест на кариотип показал 48, XXYY. Из-за этого, синдром 48, XXYY изначально считался разновидностью синдрома Клайнфелтера. Общие физические и медицинские особенности, обусловленные наличием дополнительной Х-хромосомы, включают высокий рост, развитие дефицита тестостерона в подростковом и / или зрелом возрасте (гипергонадотропный гипогонадизм) и бесплодие. Тем не менее, недавние исследования выявили некоторые важные различия у людей с кариотпом 48, XXYY по сравнению с 47, XXY[5]. Наиболее важные различия обусловлены влиянием дополнительных X и Y-хромосом на развитие нервной системы, что приводит к более высокой частоте задержек развития в раннем детстве, неспособности к обучению или умственной отсталости, трудностей с адаптивным функционированием, расстройств нервного развития, таких как СДВГ или расстройства аутистического спектра и психологические/поведенческие проблемы, включая тревогу, депрессию и нарушение регуляции настроения. Кроме того, больший процент мужчин с XXYY имеют дополнительные медицинские проблемы, такие как судороги, врожденные аномалии локтевого сустава (радиульный синостоз) и тремор по сравнению с мужчинами с XXY. XXYY до сих пор считается вариацией синдрома Клайнфелтера по некоторым определениям, главным образом потому, что патофизиология дисфункции яичек не отличается от 47, XXY, и самые последние исследования не предполагают, что должны быть какие-либо различия в оценке и лечение дефицита тестостерона в 48, XXYY по сравнению с 47, XXY[8]. Однако для психологических и поведенческих симптомов синдрома XXYY обычно требуются более обширные оценки, вмешательства и поддержка по сравнению с 47, XXY из-за более сложного вовлечения в развитие нервной системы. Наблюдается значительная вариабельность между людьми по количеству и серьезности медицинских проблем и проблем развития нервной системы, связанных с XXYY, и у некоторых людей наблюдаются легкие симптомы, в то время как другие страдают более значительным образом[2].
Примечания[править | править код]
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ 1 2 3 4 Tartaglia N, Davis S, Hench A, et al. (June 2008). “A New Look at XXYY Syndrome: Medical and Psychological Features”. Am. J. Med. Genet. A. 146A (12): 1509–22. doi:10.1002/ajmg.a.32366. PMC 3056496. PMID 18481271.
- ↑ 1 2 3 48,XXYY syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. rarediseases.info.nih.gov. Дата обращения: 9 июня 2019.
- ↑ Genetics Home Reference. 48,XXYY syndrome (англ.). Genetics Home Reference. Дата обращения: 9 июня 2019.
- ↑ 1 2 University of California – UC Newsroom | Researchers define characteristics, treatment options for XXYY Syndrome. web.archive.org (1 апреля 2010). Дата обращения: 9 июня 2019.
- ↑ Visootsak J, Rosner B, Dykens E, Tartaglia N, Graham JM, Jr (2007). “Behavioral phenotype of sex chromosome aneuploidies: 48, XXYY, 48, XXXY, and 49,XXXXY”. Am. J. Med. Genet. A. 143A (11): 1198–1203. doi:10.1002/ajmg.a.31746. PMID 17497714.
- ↑ INSERM US14– ALL RIGHTS RESERVED. Orphanet: 48,XXYY syndrome (англ.). www.orpha.net. Дата обращения: 9 июня 2019.
- ↑ 1 2 Cotran, Ramzi S.; Kumar, Vinay; Fausto, Nelson; Nelso Fausto; Robbins, Stanley L.; Abbas, Abul K. (2005). Robbins and Cotran pathologic basis of disease. St. Louis, Mo: Elsevier Saunders. p. 179. ISBN 978-0-7216-0187-8.
Источник
В большинстве случаев анеуплоидий половых хромосом (встречающихся приблизительно у 0,3% живых новорожденных) отмечается повышенный риск психиатрических отклонений. С другой стороны, анеуплоидии половых хромосом относительно часто встречаются среди детей, состоящих на учете психиатра. По результатам одного из исследований (Crandall et al., 1972) 1,6% детей, обратившихся к детскому психиатру, имели анеуплоидии половых хромосом. Данные аномалии связаны с несколько повышенным риском задержки умственного развития. Например, по результатам исследования Ratcliffe et al. (1994) было продемонстрировано, что кариотип XXY выявлялся у 5,4 на 1000 фенотипических мальчиков, наблюдавшихся в учреждениях для лиц с умственной отсталостью, а коэффициент IQ у таких детей был несколько ниже, чем в контрольной группе. Средний коэффициент IQ у девочек с тремя Х-хромосомами составил 86,8, у мальчиков с кариотипом XXY — 91,4, а у мальчиков с кариотипом XYY — 101,7; все показатели были значимо ниже, чем в контрольных группах (109,1 —у девочек и 115,8 — у мальчиков). Среди детей с дополнительной половой хромосомой также отмечалась меньшая окружность головы (даже при рождении), что отражает воздействие дополнительных половых хромосом на рост головного мозга в пре- и постнатальном периоде. Кариотип XXX был выявлен у 4,3 из 1000 фенотипических девочек, находящихся в лечебных учреждениях (Pennington et al., 1980). Средний коэффициент IQ при кариотипе ХО (синдром Тернера) находится в пределах нормы, но многие девочки с данным кариотипом путают право-лево и имеют отклонения организации восприятия, их вербальный 1Q с определенной индивидуальной вариабельностью имеет склонность превышать IQ исполнения заданий (Temple и Carney, 1993). Более серьезная умственная отсталость, очевидно, присуща пациентам со сложными аномалиями, к примеру, фенотипическим мужчинам с тремя и более Х-хромосомами (Zaleski et al., 1966) или с дупликацией обеих половых (X и Y) хромосом (Schlegel et al., 1965). Тем не менее, основные отклонения, встречающиеся у таких детей, включают сложности обучения и восприятия и поведенческие отклонения (Bender et al., 1983, Ratcliffe et al., 1994). а) XYY синдром. У мальчиков с дополнительной Y-хромосомой (около 0,1% всего мужского населения) имеется значительно повышенный риск задержки и других отклонений речевого развития, приступов гнева и агрессивности. У таких пациентов также часто отмечается гипотония и раннее появление дефицита внимания с гиперактивностью. Приступы гнева часто встречаются в раннем детском возрасте. Распространена сниженная общительность, а риск аутизма повышен (Hagerman, 1989). Показатель IQ обычно в пределах нормы, но очень часто отмечаются трудности в обучении, а частота задержки умственного развития по сравнению с общей мужской популяцией повышена. На основании как минимум одного объективного проспективного исследования предполагается небольшое повышение склонности к агрессивному поведению и проявления садизма в половой жизни во взрослом возрасте (Schiavi et al., 1988). Большинство пациентов имеет высокий рост и пропорциональное строение относительно размеров ног и головы. Пациенты обычно не менее чем на 13 см выше своего отца. Обследование на выявление кариотипа XYY необходимо у высокорослых мальчиков с отклонениями в поведении и трудностями в обучении.
б) XXY синдром (синдром Клайнефельтера). У мальчиков с одной или более дополнительной Х-хромосомой (около 0,1-0,2% всех новорожденных мальчиков) диагностируется синдром Клайнфелтера (Lanfranco et al., 2004). Почти 2/3 случаев составляет кариотип 47XXY. Окружность головы, рост и вес при рождении ниже средних параметров. Приблизительно с возраста 2-4 лет отмечается увеличение скорости роста, особенно длины ног. Средний рост пациентов во взрослом возрасте приблизительно на 13 см превышает рост отца. Размер головы не компенсируется. Дополнительная Х-хромосома ингибирует рост головного мозга во внутриутробном периоде. У мальчиков с синдромом Клайнфелтера обычно отмечается снижение вербального IQ, в то время как общий коэффициент IQ остается в пределах нормы (или слегка снижен); обычно коэффициент IQ варьирует от 60 до 130. Отмечается задержка речевого развития, и в большинстве случаев лечение по поводу отклонений развития речи начинается задолго до проведения хромосомной диагностики. Пациенты часто бывают неуклюжими и имеют отклонения типичные для детей с DAMP (дефицит внимания (Deficits in Attention), двигательного контроля (Motor control) и восприятия (Perception), иногда с тенденцией к гипоактивности. Данные отклонения могут усиливаться за счет аномального строения тела и нервно-мышечных характеристик. Нередки серьезные проблемы с чтением и письмом, не зависящие от общего IQ. Изменения на МРТ в наиболее пораженных областях (лобной, височной и моторной зонах) и менее затронутой теменной области соответствуют выявляемым когнитивным и поведенческим отклонениям у пациентов с кариотипом XXY (Giedd et al., 2007). Начиная со среднего детского возраста отмечается тенденция к удлинению ног, а размах рук часто превышает рост. Пенис может быть уменьшенного или нормального размера, яички почти всегда уменьшены, а продукция спермы нарушена. У многих пациентов отмечается увеличение молочных желез, а частота рака груди при данном заболевании выше, чем у здоровых мужчин. Часто пациенты характеризуются как застенчивые, неуверенные в себе. У большинства отмечаются легкие/умеренные проблемы социального взаимодействия и склонность избегать коллективной деятельности. У небольшого количества пациентов отмечается аутизм. Результаты одного проспективного исследования позволяют предположить, что взрослые пациенты с синдромом Клайнфелтера менее сексуально активны и более склонны к подчинению в сексуальной жизни, чем другие мужчины (Schiavi et al., 1988). Лечение тестостероном следует начинать в препубертатном периоде под наблюдением эндокринолога, являющегося специалистом по синдрому Клайнфелтера. Интрацитоплазматическая инъекция спермы позволяет осуществлять детородную функцию, даже в случае, если в эякуляте сперматозоиды отсутствуют. Даже в случае стойкой азооспермии возможно выделение сперматозоидов из биптатов яичек и реализации, беременности и деторождения (Lanfranco et al., 2004).
в) Х0 синдром (45Х0 синдром, синдром Тернера). Большинство случаев кариотипа 45X0 выявляется при рождении по характерному отеку тыла стоп и кистей и крыловидным складкам на шее. Кроме того, такие дети часто рождаются раньше срока с низкой массой тела и низким ростом. У девочек с данным синдромом частота психиатрических отклонений невысокая. Обычно 1Q в пределах нормы, но несколько снижен показатель исполнения заданий. Приблизительно у 10% пациентов отмечается определенный уровень задержки умственного развития. Зрительно-пространственные навыки часто развиты слабо, что может влиять на математические способности. По шкале детского интеллекта Векслера (WISC-R) отмечается плохое выполнение субтестов «композиции из кубиков» и «складывания фигур». Многие пациенты с синдромом Тернера гиперактивны в раннем детском возрасте, но начиная с подросткового возраста склонны к гипоактивности (Swillen et al., 1993). Предполагается взаимосвязь синдрома Х0 с нервной анорексией (в частности, при мозаичном генотипе), но результаты систематического хромосомного исследования не подтверждают данного предположения (Rastam et al., 1991). После подросткового возраста пациенты характеризуются низкорослостью и инфантильным строением наружных и внутренних половых органов. Заместительная терапия эстрогенами необходима, но, тем не менее, не существует четких рекомендаций по поводу возраста начала лечения. Многие женщины с синдромом Тернера выходят замуж и во взрослом возрасте живут нормальной жизнью. Пациентки с синдромом Тернера, получившие Х-хромосому от матери, имеют больше проблем в социальном взаимодействии, чем женщины, получившие Х-хромосому от отца (Skuse et al., 1997). г) ХХХ синдром. Девочки с кариотипом XXX составляют 0,1% новорожденных девочек. У таких детей имеется повышенный риск нарушений речи и обучения, IQ близок к нижней границе нормы, что приводит к необходимости применения специальных образовательных программ, тем не менее, нарушения чтения и письма соответствуют общему уровню IQ. Пациентки характеризуются застенчивостью, незрелостью и различными поведенческими проблемами (Linden et al., 1988). Характерен высокий рост и плохая координация движений. Данные отклонения в сочетании со странным поведением приводят к высокой частоте обращения в специализированные учреждения. – Вернуться в оглавление раздела “Неврология.” Редактор: Искандер Милевски. Дата публикации: 5.12.2018 Оглавление темы “Наследственные синдромы в неврологии.”:
|
Источник