
Врожденный синдром с изменением морфологии лейкоцитов

Врожденные нейтропении – это группа генетически детерминированных заболеваний, которые характеризуются снижением уровня нейтрофильных лейкоцитов ниже 1500/мкл, а у детей до 1 года – ниже 1000/мкл. Клинически это проявляется частными бактериальными инфекциями, задержкой в психофизическом развитии. Наиболее распространенным признаком врожденных нейтропений являются частые гингивиты и стоматиты. Диагноз выставляется на основании анамнеза, данных осмотра, общего анализа крови и миелограммы. Тактика лечения зависит от формы патологии. Для стимуляции синтеза нейтрофильных гранулоцитов применяется гранулоцитарный колониестимулирующий фактор (Г-КСФ).
Общие сведения
Врожденные нейтропении – это группа наследственных патологий, которые передаются по аутосомно-доминантному или аутосомно-рецессивному типу и проявляются уменьшением количества нейтрофилов в периферической крови. Все заболевания, входящие в эту группу, были описаны в ХХ веке: синдром Костмана – в 1956 году, семейная доброкачественная нейтропения – в 1939 году, циклическая нейтропения – в 1910 году, синдром «ленивых лейкоцитов» – в 1964 году. Встречаются данные патологии редко. Распространенность колеблется от 1-2:100000 до 1 случая на 1 млн. младенцев. Врожденными нейтропениями с одинаковой частотой болеют как мальчики, так и девочки. Прогноз зависит от формы заболевания, при синдроме Костмана летальность достигает 97-100%, в то время как при семейной доброкачественной нейтропении исход, как правило, благоприятный.

Врожденные нейтропении
Причины врожденных нейтропений
Врожденные нейтропении – это генетически обусловленные заболевания, которые наследуются по аутосомно-доминантному или аутосомно-рецессивному типу. Синдром Костмана проявляется мутацией в гене ELA2, находящемся на 19р13.3. Данный ген кодирует фермент – нейтрофильную эластазу. Точная роль ее неизвестна, но, вероятнее всего, при ее дефекте у нейтрофилов еще в костном мозге запускается процесс апоптоза. Реже данная патология может быть вызвана дефектами генов GFII и 6-CSFR, кодирующих фактор активации эластазы нейтрофилов и рецепторы к гранулоцитарно-макрофагальному колониестимулирующему фактору. Циклическая врожденная нейтропения также развивается на фоне мутации ELA2, однако, апоптоз при данной форме проходит не так интенсивно, что обеспечивает менее выраженный дефицит нейтрофилов. Эти две формы врожденных нейтропении наследуются по аутосомно-рецессивному типу.
Синдром «ленивых лейкоцитов» возникает на фоне нарушения процесса выхода гранулоцитов из костного мозга в системный кровоток. Патогенез данной формы врожденной нейтропении основывается на мутации белка, кодирующего клеточную мембрану нейтрофилов, а также их ускоренном апоптозе. При семейной доброкачественной нейтропении у детей нарушается процесс дифференциации гранулоцитов в костном мозге – нейтрофилы остаются на стадии метамиелоцитов. Также существует целый ряд врожденных синдромов, одним из проявлений которых является уменьшение количества нейтрофильных лейкоцитов. Сюда относятся синдром гипер-IgM, ретикулярная дисгенезия, синдром Чедиака-Хигаси, синдром Швахмана-Даймонда, Барта и др.
Классификация врожденных нейтропений
В педиатрии нейтропении разделяются на врожденные и приобретенные формы. К врожденным относятся:
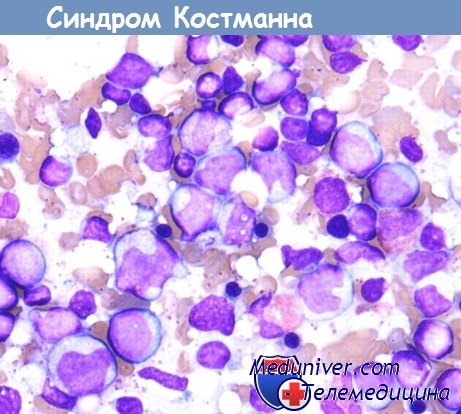
- Синдром Костмана. В основе заболевания лежит ранний апоптоз и отсутствие в периферической крови зрелых форм нейтрофильных лейкоцитов. В костном мозге определяются пролиферирующие клетки до миелоцитов. Характеризуется тяжелой клинической картиной уже на первых месяцах жизни.
- Циклическая нейтропения. Данная форма врожденной нейтропении проявляется недостаточным гранулоцитопоэзом, который имеет повторяющийся характер. Клинические симптомы возникают одновременно с эпизодами агранулоцитоза.
- Семейная доброкачественная нейтропения. В основе этой формы врожденной нейтропении лежит нарушение дозревания нейтрофильных гранулоцитов. Клинически проявляется редко. Специфического лечения не требует.
- Синдром «ленивых лейкоцитов». Суть заболевания заключается в нарушении хемотаксиса нейтрофилов. Проявляется частыми воспалительными заболеваниями уже с младенчества.
Симптомы врожденных нейтропений
Клинические картины различных форм врожденной нейтропении имеют как общие аспекты, так и определенные различия. При всех видах часто возникают воспалительные заболевания органов, тесно контактирующих с внешней средой и наиболее чувствительных к ослаблению иммунной системы. Сюда относятся кожа, слизистая оболочка рта, трахеобронхиальное дерево, легкие, внешнее и среднее ухо. Также почти всегда присутствуют астеновегетативный и интоксикационный синдромы. Однако возраст, в котором манифестирует врожденная нейтропения, частота и степень тяжести обострений могут существенно варьировать.
Синдром Костмана характеризуется выраженной симптоматикой уже в первые месяцы жизни ребенка. Первичные признаки – лихорадка неясной этиологии, частые бактериальные заболевания кожи и подкожной жировой клетчатки (фурункулы, флегмоны). У таких детей медленно заживают пупочные ранки, плохо поддается лечению омфалит. Может наблюдаться задержка в психическом и физическом развитии. Постепенно присоединяются гепатоспленомегалия и лимфаденопатия. Характерный признак врожденной нейтропении – поражение слизистых оболочек рта и десен (гингивит и стоматит). При синдроме Костмана также отмечаются рецидивирующие пневмонии, абсцессы легких, циститы, отиты, уретриты, пиелонефриты, гастродуодениты, парапроктиты, перитониты и т. д. Все перечисленные патологии склонны к генерализации, что без раннего лечения приводит к развитию сепсиса и смерти.
Циклическая нейтропения проявляется в возрасте до 1 года. Также характеризуется поражением кожи, внешнего уха, слизистых оболочек рта и десен. Типичной особенностью данной формы врожденной нейтропении является периодичность рецидивов. Обострения могут возникать каждые 14-49 дней, зачастую – каждые 3 недели. В тяжелых случаях, особенно при инфицировании анаэробной микрофлорой, развиваются тяжелые осложнения в виде перитонита и сепсиса, однако их вероятность значительно меньше, чем при синдроме Костмана. С возрастом частота и тяжесть рецидивов снижаются.
Семейная доброкачественная нейтропения проявляется в возрасте от 2-3 месяцев до 1 года. В клинической картине этой врожденной нейтропении превалируют редкие гингивиты и стоматиты, фурункулез. Еще реже встречаются отиты и поражения легких. Перечисленные заболевания, как правило, протекают в легкой форме, общее состояние ребенка нарушено мало. Клиническая картина синдрома «ленивых лейкоцитов» наблюдается уже на первых месяцах жизни. Наиболее часто у пациентов диагностируются бактериальные поражения верхних дыхательных путей (ларингиты, фарингиты, трахеиты), пневмонии, гингивиты и стоматиты.
Диагностика врожденных нейтропений
Диагностика врожденных нейтропений основывается на сборе анамнестических данных, физикальном обследовании, результатах лабораторных и инструментальных исследований. Из анамнеза педиатром или неонатологом обязательно устанавливается наличие подобных наследственных заболеваний у родителей или других родственников. Физикальное обследование может выявить умеренное отставание в физическом развитии, лимфаденопатию, гепатоспленомегалию. При развитии бактериальных осложнений будут обнаруживаться другие специфические изменения.
Основа диагностики врожденных нейтропений – общий анализ крови и миелограмма. В ОАК определяется снижение уровня лейкоцитов ниже 4,5х109/л, а нейтрофильных гранулоцитов – до 1000/мкл и ниже у грудных детей и до 1500/мкл и ниже у детей старше 1 года. Врожденные нейтропении почти всегда сопровождаются моноцитозом и эозинофилией. В зависимости от формы уровень нейтрофилов может варьировать, как и изменения в костном мозге при его пункции. В миелограмме при синдроме Костмана выявляются только клетки-предшественники нейтрофилов – миелобласты, промиелоциты, миелоциты.
Циклическая нейтропения также проявляется отсутствием зрелых форм, однако при повторных тестах может обнаруживаться лейкоцитоз. Миелограмма при семейной доброкачественной нейтропении характеризуется большим числом метамиелоцитов и дефицитом зрелых нейтрофилов. Синдром «ленивых лейкоцитов» проявляется чрезмерным насыщением костного мозга клетками всех этапов дифференциации, в том числе и зрелыми. Другие изменения в лабораторных или инструментальных тестах соответствуют возникшим осложнениям врожденных нейтропений.
Лечение врожденных нейтропений
Лечение врожденных нейтропений зависит от формы патологии. Основу терапии составляет гранулоцитарный колониестимулирующий фактор (Г-КСФ). Данный гормональный препарат стимулирует синтез и дифференциацию нейтрофильных гранулоцитов в костном мозге. При синдроме Костмана и условии отсутствия генной мутации Г-КСФ применяется пожизненно. Также при данной форме врожденной нейтропении может проводиться пересадка костного мозга. При циклической нейтропении Г-КСФ назначается за 2-3 дня до развития агранулоцитоза. Доброкачественная семейная нейтропения и синдром «ленивых лейкоцитов», как правило, не требуют использования Г-КСФ за исключением тяжелых форм.
При развитии бактериальных осложнений на фоне врожденной нейтропении осуществляется массивная антибактериальная терапия. Как правило, назначаются антибиотики широкого спектра действия – цефалоспорины III-IV поколения, макролиды. В тяжелых случаях могут применяться внутривенные иммуноглобулины. При необходимости проводят дезинтоксикационную терапию, по показаниям используют симптоматические средства.
Прогноз и профилактика врожденных нейтропений
Прогноз при разных формах врожденных нейтропений различается. При синдроме Костмана большинство детей умирают от тяжелых, резистентных к лечению бактериальных осложнений на протяжении первых месяцев жизни. Циклическая нейтропения и синдром «ленивых лейкоцитов» имеют более благоприятный прогноз – летальный исход возможен только при тяжелых вариантах течения и отсутствии своевременной диагностики. Семейная доброкачественная нейтропения в большинстве случаев не приводит к смерти ребенка – с возрастом число нейтрофилов возрастает, иммунитет стабилизируется.
Специфической профилактики врожденных нейтропений не существует. Неспецифические превентивные меры включают в себя оценку риска развития генетических мутаций у ребенка еще до его рождения путем медико-генетического консультирования. Данное обследование можно пройти у врача-генетика в специализированных центрах. Уже беременным женщинам проводят кордоцентез, амниоцентез, плаценто- или хориоцентез с последующим кариотипированием. С целью профилактики спонтанных мутаций, которые также могут сопровождаться врожденными нейтропениями, на период беременности следует полностью исключить воздействие всех тератогенных факторов на плод. В их число входят алкоголь, наркотики, табачные изделия, химикаты, ионизирующее излучение, некоторые медикаменты и др.
Источник
Генетические (наследственные) аномалии лейкоцитов — это результат хромосомных нарушений, передающихся в одной семье через множество поколений. Такие нарушения также называют генными, поскольку мутации или аномалии обусловлены изменением в генах (хромосомах) человека. Передаются они непосредственно в процессе зачатия. Оплодотворенная яйцеклетка уже сама по себе является носителем генетической мутации.
- Аномалия Мея-Хегглина
- Аномалия Альдера
- Аномалия Пельгера-Хюэта
Существует несколько видов генетических аномалий лейкоцитов. В целом генные нарушения проявляются в различных симптомах и в разном возрасте, не обязательно с рождения. Течение таких заболеваний может быть скрытым, и его наличие определяет анализ крови или генетическое исследование.
Причинами генетических аномалий лейкоцитов являются:
- факторы среды, влияющие на организм;
- лекарственные препараты;
- радиация.
Причин таких аномалий множество, в отдельно взятой семье носителем недуга может быть любой человек.
Аномалия Мея-Хегглина
Аномалия Мея-Хегглина — это редкое наследственное заболевание, характеризующееся появлением в крови аномально больших и бесформенных тромбоцитов, а также гигантских и дефектных лейкоцитов. Дефектные лейкоциты содержат так называемые островки Деле, имеющие размер от 2 до 5 мкм. Расположены они в жидкой среде клетки — цитоплазме. Природа происхождения таких островков инфекционная, при клиническом изучении было отмечено, что островки Деле реагируют на метиленовую синьку, окрашиваясь в голубой цвет, а с оксидазой в реакцию не вступают. Аномальный ген может быть унаследован от обоих родителей, а также может являться результатом новой мутации (изменения генов) у отдельных больных. Риск передачи аномального гена от родителей к потомству составляет 50% для каждой беременности, независимо от пола будущего ребенка.
Симптомы аномалии Мея-Хегглина
Проявления аномалии Мея-Хегглина у некоторых больных могут полностью отсутствовать, а у других проявляются в аномальных кровотечениях. В легких случаях лечение аномалии не требуется. В более тяжелых случаях необходимо переливание тромбоцитов.
Основные симптомы:
- красные или фиолетовые пятна на коже (пурпура);
- кровотечение из носа;
- кровотечение изо рта во время стоматологических процедур;
- головные боли;
- односторонняя слабость мышц (половины тела);
- внутричерепные кровотечения.
Кровотечения могут усиливаться после прекращения приема стероидных препаратов.
Последние исследования аномалии Мея-Хегглина показали, что заболевание входит в группу семейства пяти аутосомно-доминантных расстройств, для которых характерно присутствие в крови гигантских тромбоцитов, каждый из которых содержит различные вариации (аллели) одного и того же гена. Другие нарушения, связанные с появлением гигантских тромбоцитов, это: синдром Себастьяна, синдром Фехтнера, синдром Эпштейна, макротромбоцитопенический синдром.
Медики полагают, что понимание механизмов какого-либо из указанных выше синдромов может помочь лучше изучить этиологию аномалии Мея-Хегглина.
Лечение аномалии Мея-Хегглина
Основной метод лечения, устраняющий опасные симптомы — это переливание тромбоцитов. Легкие проявления аномалии Мея-Хегглина в лечении не нуждаются. Носители дефектного гена должны в обязательном порядке получить консультацию специалиста по генетике.
Аномалия Альдера
Аномалия Альдера — это редкое аутосомно-рецессивное заболевание, при котором в крови накапливаются белково-углеводные комплексы, называемые мукополисахаридами.
Накопление частично деградированных (разорванных) белково-углеводных комплексов в лизосомах можно определить по окрашиванию зрелых белых кровяных клеток лейкоцитов в фиолетовый цвет. Эти вкрапления можно обнаружить при анализе крови, причем располагаться они могут кластерами, а не диффузно по цитоплазме. Обнаружить такие вкрапления чаще можно в костном мозге, чем в периферической крови.
Основные физические недостатки, к которым приводит аномалия Альдера, – это карликовость и гаргоилизм (формирование неправильных, грубых, искаженных черт лица). Название «гаргоилизм» происходит от французского слова «гаргулья».
Симптомы аномалии Альдера (внешние):
- широко посаженные глаза;
- приплюснутая переносица;
- толстый язык;
- открытый рот;
- мелкие зубы.
Поражаются также суставы, кости, нервная система и некоторые внутренние органы.
Поскольку заболевание вызвано генетическими причинами, эффективноголечения в настоящее время не существует.
Аномалия Пельгера-Хюэта
Аномалия Пельгера-Хюэта — наследственное заболевание, при котором нейтрофилы имеют ядра неправильной формы, в частности, эллипса, фасоли, боба, а также серединную перемычку. Может передаваться по наследству от одного из родителей. Впервые заболевание было описано голландским гематологом Пельгером в 1928 году. Он впервые обнаружил лейкоциты с ядрами в форме гантель, а также отметил уменьшение количества ядерных сегментов в лейкоцитах и слипание ядерного хроматина.
Позднее, в 1931 году, голландский педиатр Хюэт определил данное заболевание как наследственное.
Чаще всего аномалия Пельгера-Хюэта диагностируется у больных с миелоидной лейкемией, миелодисплазией и острым лимфобластным лейкозом. Количество нейтрофилов с аномальной морфологией доходит до 50% или даже более. Это отличает настоящую аномалию Пельгера-Хюэта от её псевдо-варианта, при котором количество дефектных лейкоцитов колеблется в пределах 5%.
Симптомы аномалии Пельгера-Хюэта
Данная аномалия приводит к появлению таких внешних признаков, как:
- скелетные деформации и нарушения (полидактилия, укороченные пястные кости, укороченные верхние конечности);
- низкорослость;
- гиперкифоз (явно искривленный в одну сторону позвоночник).
В некоторых случаях аномалия Пельгера-Хюэта наблюдается в крови пациентов, перенесших химиотерапию. Как правило, она исчезает после окончания лечения, спустя примерно 10-14 дней. Данное нарушение также характерно для больных микседемой, туберкулезом, малярией, мышечной дистрофией, множественной миеломой и для людей с повышенной чувствительностью к лекарственным препаратам.
Источники статьи:
https://www.labce.com
https://vetbook.org
https://emedicine.medscape.com
https://www.webmd.com
https://rarediseases.org
https://emedicine.medscape.com
По материалам:
©2001 – 2015 LabCE.
©2015 NORD – National Organization for Rare Disorders, Inc.
©2005-2015 WebMD, LLC.
Vikramjit S Kanwar, MBBS, MBA; Chief Editor: Robert J Arceci, MD, PhD
Смотрите также:
У нас также читают:
Источник
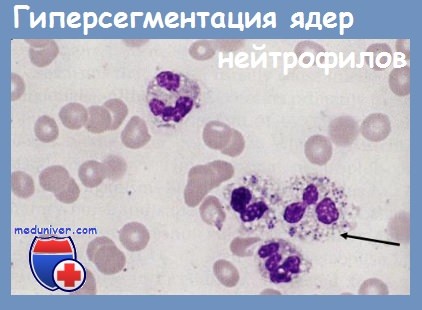
Аномалии лейкоцитов – классификация, видыЛейкоцитарные аномалии делятся на две группы: нейтрофильные гранулоцитарные аномалии и эозинофильные гранулоцитарные аномалии. Среди аномалий нейтрофильных гранулоцитов известны: Аномалия передается доминантно. Проявление носит гетерозиготный или гомозиготный характер. Аномалия является чисто гематологической; даже носители-гомозиготы не представляют какого-либо клинического проявления. Эту аномалию описал Undritz в 1943 г. Реактивное нуклеарное гиперсегментирование появляется при мегалобластических анемиях, где после исчезновения мегалобластов вследствие специфического лечения, гиперсегментирование нейтрофилов может персистировать еще долгое время. Реактивное гиперсегментирование появляется также при различных хронических анемиях, при аноксемиях, декомпенсированных кардио-пневмопатиях. Иногда, при тяжелых инфекциях, вместо отклонения «влево» лейкоцитарной формулы, появляется гиперсегментирование нейтрофилов.
2. Аномалия Давидсона (Davidson) характеризуется наследственно-конституционной полиплоидией нейтрофильных гранулоцитов (тетраплоидия). Известно, что в нормальном состоянии, двуядерные нейтрофильные гранулоциты (два отдельных ядра) существуют лишь в пропорции 0—2°/00. При аномалии Давидсона мы находим 3—16°/00 двуядерных нейтрофилов, имеющих по два диплоидных ядра, нормального вида. Аномалия передается доминантно и имеет гетерозиготный характер. Описали ее Davidson и сотр. (цитируемые Undritz). Аномалия не сопровождается никаким клиническим или биологическим проявлением. Подобное, реактивное проявление встречается при кровяных пролиферативных процессах, при токсиинфекционных процессах и после повторных применений ионизирующих излучений. 3. Наследственно-конституционная аномалия размножения в нейтрофилах специфических половых нуклеарных придатков. Нормально, у женщин 3—5% сегментированных нейтрофильных лейкоцитов имеют специфически половые нуклеарные придатки, которые обнаружили Davidson и Smith в 1954 г. При вышеупомянутой аномалии, которую открыл Undritz в 1967 г., пропорция этих придатков возрастает в среднем до 13% у гетерозиготов и до 35% в единственном случае гомозигота, известном до настоящего времени. Гетерозиготы не представляют никаких клинических или биологических проявлений. В единственном случае гомозигота констатировалась небольшая анемия, которая могла произойти и по другим причинам. Вышеописанные образования зависят от числа нуклеарных долек. 5. Аномалия Земана (Seman) соответствует наследственно-конституционному размножению неспецифических нуклеарных придатков нейтрофилов, констатированную Земаном в 1959 г. (цитированный Undritz), в одной семье, где у вообще здоровых лиц, необычайно, процент нейтрофильных гранулоцитов представлял неспецифические нуклеарные придатки („small clubs”, „sessile nodules”, „rackets” и т.д.). Передача аномалии имеет доминантный характер; проявление повидимому гетерозиготное. Клинические проявления отсутствуют. Undritz высказывает некоторые сомнения в связи с существованием этой аномалии. Впрочем, в связи с обоими аномалиями (Земана и предыдущей), существуют некоторые разногласия, на которые мы считаем целесообразным обратить внимание читателей: «специфически-половой нуклеарный придаток», который описали Davidson и Smith, был представлен первыми авторами как имеющим форму «барабанной палочки» (drum-stiks); несколько позже Kosenow и сотр. (цитированные Bessis) утверждали, что и образования, называющиеся „sessile nodules”, которые другие авторы, во главе с Undritz (1974), считают неспецифическими для женского пола, являются все-таки специфическими и равнозначными с „drum-sticks” в определении пола. С другой стороны, в связи с аномалией, описанной Seman, никто не упомянул о том, какая пропорция нуклеарных придатков является нормальной и где начинается аномалия. Так, мы находим у многих больных разные формы придатков в очень высокой пропорции, то же самое и у нормальных лиц, так что мы не располагаем сколько-нибудь определенным критерием, который позволил бы утверждать, что мы имеем дело с аномалией. В наше описание мы включили обе аномалии, так как они фигурируют во всех трудах по гематологии, но с упомянутыми оговорками.
6. Аномалия Костмана (Kostmann) — наследственно-конституционный анейтроцитоз, доходящий до полного отсутствия нейтрофильных гранулоцитов в периферической крови, причем эозинофилы и базофилы присутствуют в нормальной пропорции. Лейкоцитарная формула показывает очень высокий лимфомоноцитоз. Аномалия передается рецессивно; ее проявление наблюдается только у гомозигота. Kostmann (цитированный Undritz) обнаружил эту аномалию у 14 грудных детей, которые принадлежали родственным семьям, проживавшим на очень ограниченной территории в Швеции. Другие случаи не отмечались. Аномалия сопровождается такой симптоматологией, которую можно считать естественным последствием анейтроцитоз а: различные инфекционные локализации (фурункулы, абсцессы, флегмоны), с лихорадкой и фатальной эволюцией в течение максимум одного года. До сих пор не был найден никакой эффективный антибиотик. Гетерозиготные родители асимптоматичны. Дифференциальная диагностика производится по отношению ко всем токсиинфекционным, аллергическим, иммунологическим, туморальным и пр. «агранулоцитозам».
7. Аномалия Гицига (Hitzig), считается более доброкачественной формой предыдущей аномалии. Речь идет о семейной нейтропении, сопровождающейся гипергаммаглобулинемией, передающейся доминантно, с гетерозиготным проявлением. Undritz считает, что гомозиготы не были обнаружены, потому что они нежизнеспособны. Эту аномалию описал Hitzig в 1959 г. У гетерозиготов инфекционные осложнения нелетальны. Дифференциальная диагностика производится по отношению ко всем нейтропениям, которые встречаются в практике. Среди аномалий эозинофильных гранулоцитов известны: Реактивное гиперсегментирование эозинофилов встречается при аллергиях, пернициозной анемии, болезни Годжкина и при тропической эозинофилии. 2. Аномалия Презентей (Presentey) соответствует наследственно-конституционному дефициту пероксидаз в эозинофилъных гранулоцитах. В нейтрофилах и моноцитах, реакция остается позитивной (Undritz). С генетической точки зрения, способ передачи еще не выяснен. Согласно Presentey, передача происходит рецессивно; Undritz, наоборот, утверждает, что эта аномалия передается доминантно. Проявление гетерозиготное. Не существует никаких клинических проявлений. Дифференциальная диагностика производится с отсутствием пероксидаз благодаря порочной технике. – Также рекомендуем “Аномалии лимфоцитов – семейная амавротическая идиотия” Оглавление темы “Гематология”:
|
Источник