
Смита лемли опитца синдром с

Что такое синдром Смита-Лемли-Опица?
Синдром Смита-Лемли-Опица (сокр. ССЛО, дефицит 7-дегидрохолестерол редуктазы) — это изменчивое генетическое заболевание, которое характеризуется медленным ростом до и после рождения, маленькой головой (микроцефалия), умственной отсталостью от легкой до умеренной степени и множественными врожденными дефектами, включая определенные черты лица, волчью пасть, пороки сердца, сросшиеся пальцы второго и третьего пальцев, лишние пальцы рук и ног и недоразвитые наружные гениталии у мужчин. Тяжесть ССЛО сильно различается у больных, даже среди одной семье, а некоторые имеют нормальное развитие и только незначительные врожденные дефекты. ССЛО вызывается дефицитом фермента 7-дегидрохолестерин редуктазы, что приводит к нарушению метаболизма холестерина. Заболевание передается по наследству как аутосомно-рецессивное генетическое расстройство.
Признаки и симптомы
Симптомы синдрома Смита-Лемли-Опица сильно различаются у больных, но типичный паттерн аномалий включает:
- задержку роста;
- микроцефалию;
- лишние пальцы рук и ног;
- сращение второго и третьего пальцев ног;
- расщелину неба;
- недоразвитие наружных половых органов у мужчин;
- умственную отсталость.
Характерные черты лица, связанные с дефицитом фермента 7-дегидрохолестерин редуктазы, включают опущенные веки, складки во внутренних уголках глаз, мелкие морщинки на коже верхних и нижних век, ноздри, повернутые вперед, длинную верхнюю губу, перевернутую V-образную форму верхней губы, маленькую челюсть и большие внешние уши. Иногда присутствуют патологии десны, и у некоторых пациентов наблюдаются различные нарушения зрения, включая катаракту.
Случайные обнаруженные признаки включают:
- эпилепсию;
- пороки сердца;
- низкий мышечный тонус (гипотония) у детей младшего возраста;
- сужение дистального отверстия желудка (стеноз привратника);
- непроходимость кишечника.
Многие больные с синдромом Смита-Лемли-Опица имеют необычную болезненную чувствительность глаз к свету (светобоязнь).
Причины и факторы риска
ССЛО вызывается дефицитом фермента 7-дегидрохолестерин редуктазы. Дефицит фермента возникает в результате мутации гена DHCR7, унаследованного от каждого родителя.
Синдром Смита-Лемли-Опица — это аутосомно-рецессивное генетическое заболевание. Рецессивные генетические расстройства возникают, когда человек наследует один и тот же аномальный ген одного и того же признака от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, он будет носителем болезни, но обычно не бессимптомным. Риск для двух родителей-носителей передать дефектный ген и, следовательно, иметь больного ребенка, составляет 25% при каждой беременности. Риск иметь ребенка, который будет носителем, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей и будет генетически нормальным по данному признаку, составляет 25%. Риск одинаков для мужчин и женщин.
Все люди несут от 4 до 5 аномальных генов. Родители, которые являются близкими родственниками (кровными родственниками), имеют больше рисков, чем неродственные родители, иметь один и тот же аномальный ген, что увеличивает риск рождения детей с рецессивным генетическим заболеванием.
Затронутые группы населения
Распространенность дефицита фермента 7-дегидрохолестерин редуктазы при рождении оценивается примерно в 1 на 20 000–60 000 живорождений. Прогнозируемая распространенность, основанная на скрининге новорожденных на наличие носителей гена, оценивается от 1 на 1590 до 13 500, и это расхождение может быть связано с тем, что многие плоды с ССЛО рождаются мертворожденными. Это состояние одинаково встречается у мужчин и женщин, но у женщин часто не диагностируется, поскольку не видны генитальные аномалии. ССЛО чаще встречается у лиц европейского происхождения.
Диагностика
Диагноз ССЛО основан на физических признаках и обнаружении повышенной концентрации 7-дегидрохолестерина (7-DHC) в сыворотке крови или повышенного отношения 7-дегидрохолестерина к холестерину. Молекулярно-генетическое тестирование на мутации гена DHCR7 доступно и в основном используется для тестирования носителей и пренатальной диагностики.
Стандартные методы лечения
Медицинское лечение синдрома Смита-Лемли-Опица основано на конкретных проблемах, имеющихся у пострадавшего ребенка. Важно, чтобы ребенок осматривался по целому ряду заболеваний, связанных с ССЛО, включая заболевания глаз, сердца, опорно-двигательного аппарата, мочеполовой системы и желудочно-кишечного тракта, и чтобы за уходом следил врач-специалист, знакомый с дефицитом фермента 7-дегидрохолестерин редуктазы. Больным с тяжелыми заболеваниями может потребоваться операция по исправлению волчьей пасти, пороков сердца и генитальных аномалий. Добавки холестерина (один или два яичных желтка), иногда в сочетании с желчными кислотами, по-видимому, улучшают рост и снижают светочувствительность у людей с ССЛО без вредных побочных эффектов.
Родителям пострадавшего ребенка рекомендуется генетическое консультирование.
Прогноз
Прогноз зависит от тяжести заболевания и связанных с ним пороков развития. Заболевания сердца и пороки развития головного мозга могут быть смертельными. Некоторые пациенты доживают до взрослой жизни. Больные с легкой степенью поражения могут жить и работать в условиях дома.
Осложнения
Распознается множество возможных осложнений. Практически каждая клетка тела зависит от холестерина для поддержания нормальной функции; следовательно, дефицит холестерина у пациентов с синдромом Смита-Лемли-Опица может поражать каждый орган.
Те, кто наиболее серьезно страдает синдромом, либо выкидываются самопроизвольно (выкидыш), либо умирают в неонатальном периоде, несмотря на максимальную терапию.
Выжившие больные могут иметь почечную недостаточность, надпочечниковую недостаточность, эпилепсию, задержку развития и дисфункцию печени.
Источник
Синдром Смита-Лемли-Опитца – этиология, клиника, диагностикаСиндром Смита-Лемли-Опитца недавно стал объектом пристального внимания неврологов и психиатров из-за отчетливой взаимосвязи с аутистическими проявлениями. Возможно, данное заболевание имеет генетическую природу и тесно связано с аутистическими симптомами (см. ниже). Синдром Смита-Лемли-Опитца (ССЛО) регистрируется с частотой около 1 на 70000 новорожденных. Характерна высокая перинатальная смертность и высокая частота мертворождений. Среди выживших детей не менее чем у 80% отмечаются тяжелые отклонения, а у оставшихся 20% изменения фенотипа выражены умеренно. Синдром Смита-Лемли-Опитца (ССЛО) до сих пор диагностируется на основании типичных клинических признаков. Заболевание вызвано врожденным нарушением постскваленового синтеза холестерина. Нарушение синтеза холестерина при ССЛО вызвано наследственной мутацией гена 3-b-гидроксистерол-d-7 редуктазы (DHCR7). Повреждение гена DHCR7 приводит к нарушению синтеза холестерина и десмостерола, приводящее к повышению уровня 7DHC/8DHC, характерному снижению уровня холестерина и, что особенно важно, дисморфическому развитию. Открытие синдрома Смита-Лемли-Опитца вызвало новые вопросы о роли путей биосинтеза холестерина в развитии человека. В настоящее время у 250 пациентов с синдромом Смита-Лемли-Опитца идентифицирована 121 мутация; разнообразие мутаций обеспечивает различную клиническую выраженность. Для воспроизведения некоторых аномалий развития, характерных для ССЛО, и выяснения патогенеза заболевания, было создано две генетических модели синдрома на мышах. Наиболее характерными проявлениями синдрома наряду с задержкой умственного развития, аутизмом, другими поведенческими нарушениями и гипотонией является необычное строение лица с высоким плоским лбом, вывернутыми вперед ноздрями и микрогнатией, а также аномалии гениталий у мальчиков, включая крипторхизм и (или) гипоспадию, а в крайних случаях—неразвитые мужские гениталии, несмотря на нормальный кариотип XY (Scarbrough et al., 1986). Другие проявления включают аномальное строение ушных раковин, птоз век, эпикант, поперечное возвышение ладони и различные аномалии конечностей и внутренних органов (Opitz et al., 1994). У некоторых пациентов отмечаются припадки, достаточно часто встречается отсутствие аппетита. В течение первого года жизни смертность высокая. Аномалии головного мозга могут включать микроцефалию, гипоплазию лобных долей, аномальное строение извилин и гипоплазию мозжечка. Случаи наиболее выраженного поражения иногда выделяются как ССЛО 2 типа (Curry et al., 1987). Синдром наследуется аутосомно-рецессивным путем. Явное повышение частоты заболевания у мужчин, вероятно, связано с более легкой диагностикой на основании очевидных аномалий половых органов. Концентрация холестерина в плазме очень низкая, в то время как содержание 7-гидрохолестерола повышено, что предполагает дефект фермента, приводящего к разрыву двойной связи гидрохолестерола (Tint et al., 1994). Выживаемость строго коррелирует с более высоким уровнем холестерина плазмы (Tint et al., 1995). Возможна пренатальная диагностика путем определения содержания 7-дегидрохолестерола в амниотической жидкости (Dallaire et al., 1995). Потребление большого количества холестерина может нормализовать его уровень в крови, но необязательно будет клинически эффективно (Ullrich et al., 1996). Нарушения аутистического спектра часто регистрируются у пациентов с синдромом Смита-Лемли-Опитца (Gillberg и Coleman, 2000; Tierney et al., 2001; Sikora et al, 2006). Данные результаты и сообщение о низком уровне холестерина при различных случаях семейного аутизма позволяют предположить, что стериновый обмен может иметь важное значение для подгруппы пациентов с поведенческим синдромом аутизма и что хотя бы теоретически возможно «лечение» холестерином в случаях аутизма с низким уровнем холестерина. – Также рекомендуем “Синдром Смита-Магениса – этиология, клиника, диагностика” Редактор: Искандер Милевски. Дата публикации: 6.12.2018 Оглавление темы “Наследственные синдромы в неврологии.”:
|
Источник
Синдром Смита — Лемли — Опица (синдром SLOS, RSH или дефицит 7-дегидрохолестерол редуктазы) – наследственное заболевание, которое связано с нарушением метаболизма холестерина. Проявляется черепно-лицевыми аномалиями, умственной отсталостью, шестипалостью и другими врожденными неврологическими нарушениями и аномалиями развития.
Общие сведения
В 1964 г. обнаруженный у трех пациентов синдром совместно с генетиком Дж. Опицем описали американский педиатр Д. Смит и бельгийский педиатр Л. Лемли, поэтому заболевание назвали в их честь.
Детальное описание синдрома, сделанное в 1969 г., принадлежит Р. Н. Файн с соавторами – для названия данного синдрома в этом описании использованы первые буквы фамилий первых выявленных больных (синдром RSH).
Заболеванию больше подвержены новорожденные европеоидной расы – частота составляет 1:20 000 – 1:30 000 случаев. С меньшей частотой синдром встречается у представителей азиатской и негроидной расы.
Носителем мутантного гена является каждый тридцатый человек.
Заболеванию больше подвержены мальчики – соотношение со случаями заболевания у девочек составляет 3 к 1.
Формы
В настоящее время некоторыми исследователями выделяются два типа синдрома SLOS:
- к I типу относят заболевание со множественными пороками развития, которое совместимо с жизнью;
- ко II типу относят заболевание, которому свойственен ранний летальный исход.
Разница между данными формами заключается в основном в характере мутации, а не в генетической гетерогенности.
Причины развития
Синдром Смита – Лемли – Опица вызывается мутациями гена DHCR7, который расположен на 11 в регионе q13. 4.
Ген DHCR7 кодирует 7-стеролредуктазу, необходимую для биосинтеза фермента 7-дегидрохолестерол редуктазы. Этот фермент отвечает за конечную стадию производства холестерина – жироподобного воскового вещества, которое производится в организме (80%) и поступает с пищей (20%).
Холестерин:
- при взаимодействии с белками в период эмбрионального развития контролирует раннее развитие мозга, половых органов и конечностей;
- является структурным компонентом клеточных мембран и миелиновых оболочек, которые изолируют нервные клетки;
- участвует в образовании желчных кислот и выработке стероидных гормонов;
- участвует в синтезе витамина D.
В настоящее время у состоящего из 9-ти экзонов (кодирующих синтез белка и несущих наследственную информацию участков) гена DHCR7 выявлено более 120 мутаций, которые вызывают синдром SLOS. Встречаются:
- точечная замена, при которой одно азотистое основание заменяется другим;
- делеции (утрата) небольших участков гена;
- сплайсинговые мутации, при которых образуется ненормальный тип кодирующей наследственную информацию РНК.
Чаще всего мутации наблюдаются на участках 4,6,7,9 – именно с этими экзонами связано до 70% выявленных гетерогенных мутаций, причем 50% случаев заболевания вызвано четырьмя наиболее распространенными мутациями:
- Мутацией сплайсинга IVS8-1 G’-‘C, которая обнаружена в 13 мутантных аллелях. Заключается в изменении последовательности нуклеотидов в ДНК. Вызывает тяжелую форму заболевания, а ее гомозиготная форма – ранний летальный исход. Встречается в 30 % случаев заболевания у лиц европейского происхождения.
- Точечной миссенс-мутацией T93M, распространенной у лиц средиземноморского происхождения. Заключается в мутационном изменении нуклеотидной последовательности ДНК, при котором кодирующий тринуклеотид заменяет аминокислоту треонин в белке на аминокислоту метионин. В результате в плазме крови повышается уровень 7-дегидрохолестерина. Заболевание протекает в легкой форме.
- Мутацией p.W151X, которая наблюдается в 10 мутантных аллелях гена DHCR7. При гомозиготности нонсенс-мутация W151X вызывает летальную форму синдрома SLOS. Заключается в утрате способности кодона (единицы генетического кода) кодировать определенную аминокислоту, благодаря чему белок приобретает новые свойства, инактивируется или деградирует.
- Мутацией c.964-1GC.
Большинство выявленных мутаций гена DHCR7 являются точечными миссенс-мутациями, изменяющими одиночные аминокислоты и снижающими выработку холестерина.
Остальные выявленные мутации являются делециями или инсерциями (вставками) нуклеотидов, которые приводят к производству аномального фермента, устраняя его активность. Без активной 7-дегидрохолестерол редуктазы клетки не способны производить холестерин в достаточном количестве.
В результате в крови и тканях накапливаются потенциально токсичные продукты производства холестерина (уровень 7-дегидрохолестерола повышается в 2000 раз). Сочетание низкого уровня холестерина и накопление сопутствующих веществ вызывает нарушения во многих системах организма.
Тяжесть заболевания зависит от локализации и типа мутации в гене DHCR7.
Болезнь относится к аутосомно-рецессивному типу наследования.
Патогенез
Патогенез заболевания до конца не установлен, но известно, что снижение синтеза 7-дегидрохолестерол редуктазы вызывает существенное повышение в плазме уровня 7-дегидрохолестерола и дефицит холестерина.
Проведенные на крысах эксперименты показали, что избыток 7-дегидрохолестерола в мозге приводит к нарушению процесса обучения, а накопление в эмбрионах оксигенированного 7-дегидрохолестерола вызывает задержку роста.
Недостаток 7-дегидрохолестерол редуктазы вызывает у новорожденных голопрозэнцефалию (неполное разделение эмбрионального переднего мозга), отсутствие гипофиза и аномалии конечностей и гениталий.
Симптомы
Симптоматика синдрома Смита – Лемли – Опитца отличается разнообразием. Легкая форма заболевания сопровождается незначительными умственными и физическими нарушениями, а тяжелые случаи отличаются выраженными физическими пороками развития и летальным исходом в перинатальном возрасте.
Синдром SLOS в 100% случаев отличается:
- низкой массой и длинной тела при рождении;
- узким лбом;
- птозом (опущением век);
- наличием эпикантуса;
- косоглазием;
- микроцефалией и различными деформациями черепа;
- низко расположенными ушными раковинами;
- коротким носом с открытыми ноздрями и широким концом;
- недоразвитием челюсти (микрогнатией) и ее широким альвеолярным краем.
Наблюдается также:
- неправильное строение внешних половых органов – гипоспадия (неправильно расположенное отверстие мочеиспускательного канала, дисплазия крайней плоти, искривление полового члена), крипторхизм (неопущение яичка в мошонку), гипертрофия клитора, невыраженность половых органов;
- односторонняя или двусторонняя постаксиальная полидактилия (шестипалость) кистей или стоп;
- наличие поперечной складки на ладонях;
- наличие Y-образной синдактилии (сращения) II–III пальцев;
- укороченный III палец на руках;
- вальгусная стопа.
Встречаются также другие виды синдактилии или отсутствие (недоразвитие) одного или нескольких пальцев, вывих бедра, кифоз, сколиоз, наличие шейных ребер, тонкие ребра или их отсутствие.
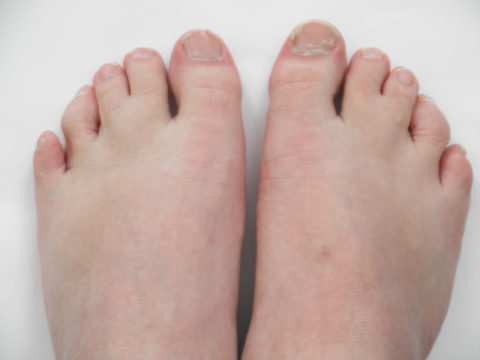 Синдактилия пальцев ног при синдроме Смита — Лемли — Опица
Синдактилия пальцев ног при синдроме Смита — Лемли — Опица
Синдром SLOS во многих случаях сопровождается:
- врожденными пороками сердца;
- аномалиями почек;
- эпилепсией;
- тугоухостью и катарактой;
- увеличением или уменьшением долей легкого;
- сужением привратника (отдел желудка);
- гастроэзофагеальной рефлюксной болезнью;
- аномалией развития толстой кишки;
- голопрозэнцефалией;
- наличием расщелины неба;
- тяжелыми нарушениями сна;
- повышенной возбудимостью, проявлениями агрессии;
- пониженным мышечным тонусом.
В большинстве случаев заболевание сопровождается отставанием в умственном развитии, а у половины больных диагностируют наличие аутизма.
К симптомам перинатального периода относят:
- воротниковый отек (больше чем 3 мм) в I триместре;
- специфические деформации у плода, которые обнаружены во II триместре при помощи УЗИ;
- маловодие.
Диагностика
Для постановки диагноза заболевания используются:
- Клинические данные.
- Данные биохимического анализа крови. Определение уровня холестерина в крови не является достоверным методом (у 10 % больных этот уровень соответствует норме), поэтому используется определение уровня 7-дегидрохолестерола в тканях или в плазме крови.
В сомнительных случаях диагноз подтверждается ДНК-диагностикой, анализом фермента 7-дегидрохолестерин редуктазы или анализом биосинтеза стерина в культуре клеток.
Пренатальная диагностика включает:
- биопсию ворсинчатого хориона, которая проводится на 10-й полной неделе вынашивания и позволяет определить уровень стеролов в ворсинах хориона;
- амниоцентез, при котором определяют уровень 7-дегидрохолестерина в амниотической жидкости;
- кордоцентез, который проводится не раньше 18 полных недель вынашивания и позволяет исследовать уровень холестерина в пуповинной крови.
Возможна также диагностика по УЗИ при характерных пороках развития плода.
Лечение
Эффективного лечения синдрома Смита – Лемли – Опитца в настоящее время не существует.
Проводится:
- Хирургическое лечение аномалий развития (дефектов конечностей и др.).
- Поддерживающая терапия, которая заключается в использовании пищевого холестерина. Пищевой холестерин поступает в организм больного либо в виде продуктов с богатым содержанием холестерина (яичных желтков и др.), либо в виде водной или масляной суспензии, либо в кристаллической форме. Обычно препаратом выбора является порошок Cholesterol Module, позволяющий точно рассчитывать дозу и свести к минимуму негативные последствия холестериновой диеты (запоры, рвота).
Источник