
Анализ крови при синдроме беквита видемана

Синдром Беквитта-Видемана: причины, диагностика, лечениеЭтиология и встречаемость синдрома Беквитта-Видемана. Синдром Беквитта-Видемана (MIM №130650) — панэтнический синдром, обычно спорадический, но иногда может наследоваться как аутосомно-доминантный. Синдром Беквитта-Видемана встречается приблизительно у одного из 13 700 живорожденных. Синдром Беквитта-Видемана вызван нарушением баланса экспрессии импринтированных генов в регионе р15 хромосомы 11. Эти гены включают транскрибируемые, но не транслируемые KCNQOT1 и Н19, и кодирующие белки гены CDKN1C и IGF2. В норме эти гены импринтированы и экспрессируются только из отцовского (IGF2 и KCNQOT1) или только материнского аллеля (HI9 и CDKN1C). IGF2 кодирует инсулиноподобный фактор роста, стимулирующий рост; CDKN1C кодирует супрессор клеточного цикла, ограничивающий деление и рост клеток. Транскрипция РНК Н19 и KCNQOT1 подавляет экспрессию материнской копии IGF2 и отцовской копии CDKN1С соответственно. Несбалансированная экспрессия импринтированных генов в 11р15 может происходить по множеству механизмов. Мутации в материнском аллеле CDKN1C обнаруживают в 5-10% спорадических случаев и в 40% семей с аутосомно-доминантным синдромом Беквитта-Видемана. Большинство пациентов с синдром Беквитта-Видемана, тем не менее, имеют снижение экспрессии материнского аллеля гена CDKN1С вследствие аномального импринтинга, а не мутации. У 10-20% индивидуумов с синдромом Беквитта-Видемана снижение экспрессии материнской копии CDKN1C и повышение экспрессии IGF2 вызвано отцовской изодисомией 11р15. Поскольку соматическая рекомбинация, ведущая к сегментной однородительской дисомии, происходит после зачатия, больные с сегментной однородительской дисомией — мозаики, и для выявления изодисомии может потребоваться исследование других тканей, кроме крови. Еще 1-2% больных с синдром Беквитта-Видемана имеют цитогенетически обнаруживаемую хромосомную аномалию, например, материнскую транслокацию, инверсию хромосомы 11 или дупликацию отцовской хромосомы 11р15. Таким образом, чтобы исключить структурную аномалию 11р15 при проведении генетического консультирования, необходимо кариотипирование родителей. При синдроме Беквитта-Видемана также обнаружены редкие микроделеции в гене KCNQOT1 или Н19, нарушающие импринтинг. У остальных пациентов аномалии в импринтинге и экспрессии генов остаются невыясненными.
Патогенез синдрома Беквитта-ВидеманаВ ходе гаметогенеза и в раннем зародышевом развитии у мужчин и женщин устанавливаются различные типы метилирования ДНК в генах KCNQOT1 и Н19. Аномальный импринтинг при синдроме Беквитта-Видемана легче всего обнаружить при анализе метилирования ДНК в специфических участках CpG в генах KCNQOT1 и Н19. У 60% лиц с синдромом Беквитта-Видемана обнаруживают гипометилирование материнского аллеля KCNQOT1. У других 2-7% больных гиперметилирование материнского гена Н19 снижает его экспрессию, что приводит к избыточной экспрессии IGF2. Несоответствующая экспрессия обоих родительских аллелей IGF2 может объяснить избыточный рост, наблюдаемый при синдроме Беквитта-Видемана. Аналогично снижение экспрессии материнской копии CDKN1С удаляет ограничение роста плода. Фенотип и развитие синдрома Беквитта-ВидеманаСиндром Беквитта-Видемана связан с пренатальным и постнатальным избыточным ростом. До 50% больных рождаются недоношенными и превышают массу тела, соответствующую гестационному сроку при рождении. Плацента также увеличена, а беременность часто осложняется многоводием. Кроме этого, у новорожденных с синдром Беквитта-Видемана часто бывают такие осложнения, как омфалоцеле, макроглоссия, неонатальная гипогликемия и кардиомиопатия, приводящие к 20% смертности. Неонатальная гипогликемия обычно мягкая и непостоянная, но есть сообщения о некоторых случаях более серьезной гипогликемии. Пороки развития почек и повышение кальция в моче с развитием нефрокальциноза и мочевых камней отмечают почти у половины больных. Гиперплазия различных сегментов тела или отдельных органов может выявляться уже при рождении и становится более или менее заметной со временем. Развитие больных обычно нормальное, если у них нет несбалансированной хромосомной аномалии. Дети с синдромом Беквитта-Видемана имеют повышенный риск развития эмбриональных опухолей, особенно опухоли Вильмса и гепатобластомы. Общий риск новообразований у детей с синдромом Беквитта-Видемана приблизительно 7,5%; риск значительно снижается после достижения детьми 8 лет. Особенности фенотипических проявлений синдрома Беквитта-Видемана: Лечение синдрома Беквитта-ВидеманаОказание помощи детям с синдромом Беквитта-Видемана включает лечение имеющихся симптомов, например коррекцию омфалоцеле и гипогликемии. Макроглоссия может потребовать специальных методов вскармливания или занятий с логопедом. При крупных дефектах брюшной стенки, асимметрии длины ног и при пороках развития почек может оказаться необходимым хирургическое вмешательство. Если имеется гиперкальциурия, может быть назначена терапия, направленная на уменьшение выделения кальция. Важно периодическое обследование на эмбриональные опухоли, поскольку они отличаются быстрым ростом и злокачественностью. Текущие рекомендации для исключения опухолей — УЗИ брюшной полости каждые 3 мес в течение первых 8 лет жизни и измерение сывороточного АФП каждые 6 нед в течение первых нескольких лет жизни.
Риск повторения – наследования синдрома Беквитта-ВидеманаРиск повторения для сибсов и потомства детей с синдромом Беквитта-Видемана существенно изменяется в зависимости от молекулярной основы заболевания. См. таблицу риска повторения для различных молекулярных изменений. Повышение риска синдрома Беквитта-Видемана при применении вспомогательных репродуктивных технологийВспомогательные репродуктивные технологии, например ЭКО и ИКСИ, становятся обычной процедурой, составляющей теперь во многих странах до 1-2% всех рождений. Ретроспективные исследования показали, что при беременностях, закончившихся новорожденными с синдромом Беквитта-Видемана, ЭКО использовалось в 10-20 раз чаще по сравнению с контролем. Риск синдрома Беквитта-Видемана после ЭКО оценивают как 1 на 4000, что в 9 раз выше, чем в общей популяции. Причина повышенной встречаемости дефектов импринтинга после ЭКО неизвестна. Встречаемость синдрома Прадера-Вилли, дефекта отцовского импринтинга, после ЭКО не повышена, а частота синдрома Ангельмана, дефекта материнского импринтинга, после ЭКО повышается, что позволяет предположить специфические отношения между ЭКО и материнским импринтингом. Поскольку отцовский импринтинг происходит задолго до ЭКО, а материнский происходит значительно ближе ко времени оплодотворения, роль ЭКО, как предрасполагающего к дефектам импринтинга фактора, требует серьезного анализа. Пример синдрома Беквитта-Видемана. А.Б., 27-летняя беременная, обратилась в пренатальный диагностический центр для проведения ультрасонографии 2-го уровня и генетического консультирования после планового УЗИ, обнаружившего крупный для данного гестационного возраста мужской плод с возможным омфалоцеле. Беременность, первая у каждого из родителей, наступила самопроизвольно, без вспомогательных репродуктивных технологий. После обследования ультрасонографией 2-го уровня семье выдано заключение, что плод имеет множество аномалий, наиболее соответствующих диагнозу синдрома Беквитта-Видемана, хотя не исключены и другие врожденные дефекты. Семейная пара решила не подвергаться процедуре амниоцентеза. Младенец родился кесаревым сечением в 37 нед с массой тела при рождении 9 фунтов и 2 унции (4 кг 140 г) и заметно увеличенной плацентой. Отмечены омфалоцеле, макроглоссия и вертикальные складки на мочках ушей. Консультант-генетик поставил клинический диагноз синдрома Беквитта-Видемана. После развития гипогликемии ребенок переведен в палату интенсивного наблюдения и в течение 1 нед получал внутривенные вливания глюкозы; гипогликемия разрешилась спонтанно. Результаты оценки сердечно-сосудистой деятельности нормальные, омфалоцеле откорректировано хирургическим путем без осложнений. Исследование метилирования гена KCNQOT1 подтвердило дефект импринтинга в 11р15, соответствующий диагнозу синдрома Беквитта-Видемана. Для исключения опухоли Вильмса рекомендовано каждые 3 мес УЗИ органов брюшной полости до достижения 8 лет и определение сывороточного АФП каждые 6 нед, как скрининговое обследование на гепатобластому, в течение первых 3 лет жизни. При последующих визитах, с учетом отрицательного семейного анамнеза и нормальных кариотипов родителей, дефект импринтинга в этой семье был расценен как спорадический случай синдрома Беквитта-Видемана с низким риском повторения. – Также рекомендуем “Наследственный рак молочной железы и яичников: причины, диагностика, лечение” Оглавление темы “Наследственные болезни”:
|

Источник
Медицинский эксперт статьи
х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Беквит-Видемана синдром (BWS) врожденное заболевание, которое характеризуется чрезмерно быстрым ростом в младшем возрасте, ассиметрией развития тела, повышенным риском развития рака и некоторых врожденных дефектов, нарушением поведения ребенка. Впервые описано как семейная форма омфалоцеле с макроглоссией в 1964 году, немецким доктором Ханс-Рудольф Видеманом. В 1969 году, Дж. Брюс Беквит из Университета Лома Линда, штат Калифорния, описал подобные симптомы у нескольких пациентов. Первоначально, профессор Видеман ввел термин синдром EMG, чтобы описать сочетание врожденной пупочной грыжии, макроглоссии и гигантизма. Со временем, эта патология была переименована в синдром Беквит-Видемана (BWS).
[1], [2], [3], [4]
Код по МКБ-10
Q87.3 Синдромы врожденных аномалий, проявляющиеся избыточным ростом [гигантизмом] на ранних этапах развития
Эпидемиология
Синдром Беквита-Видеманна встречается с частотой 1 на 13 700 новорождённых.
[5], [6], [7], [8]
Причины синдрома Беквита-Видеманна
Синдром Беквита-Видеманна со сложным типом наследования, локус заболевания расположен на коротком плече хромосомы 11 (CDKN1C, H19, IGF2, и KCNQ1OT1 гены). Аномальное метилирование нарушает регуляцию этих генов, что приводит к чрезмерно быстрому росту и другим характерным особенностей синдрома Беквит-Видемана.
Около 1% всех людей с данным синдромом имеют хромосомные аномалии, такие как перегруппировки (транслокации), ненормальное копирование (дублирование), или утраты (удаления) генетического материала из хромосомы 11.
Возможна молекулярно-генетическая верификация изменений этого локуса.
[9], [10], [11], [12], [13]
Симптомы синдрома Беквита-Видеманна
Заболевание характеризуется преждевременным быстрым ростом ребенка в раннем возрасте. После 8 лет рост замедляется. У некоторых детей с синдромом Беквит-Видемана отдельные части тела с одной стороны могут вырасти до аномально больших размеров (так называемая гемигиперплазия), что приводит к асимметричности внешнего вида.
Некоторые младенцы с синдромом Беквит-Видемана имеют аномально большой язык (макроглоссия), что иногда затрудняет дыхание и глотание, аномально большие органы брюшной полости (спланхномегалия), кожные складки или ямки возле ушей, гипогликемию и аномалии почек.
Дети имеют повышенный риск развития нескольких типов раковых опухолей, в частности, рака почки, опухоли Вильмса и гепатобластомы.
[14], [15], [16]
Осложнения и последствия
Возможные осложнения у больных с синдромом Беквита-Видеманна:
- вероятность неонатальной гипогликемии (60%) с развитием судорог, обусловленных транзиторным гиперинсулинизмом;
- высокая частота (10-40%) эмбриональных опухолей, особенно при нефромегалии или соматической асимметрии тела, требует наблюдения и проведения ультразвукового исследования почек 3 раза в год до 3-летнего возраста и в последующем 2 раза в год до 14-летнего возраста (своевременная диагностика опухоли Вильмса).
[17], [18], [19], [20], [21]
Диагностика синдрома Беквита-Видеманна
Диагноз синдром Беквита-Видеманна необходимо рассматривать у детей с аномалиями передней стенки живота (эмбриональной или пупочной грыжей, расхождением прямых мышц), макроглоссией, неонатальной гипогликемией и опухолями (нейробластомой, опухолью Вильмса, карциномой печени).
Диагностические критерии:
- Большая масса тела при рождении или постнатальное опережение физического развития.
- Дефекты закрытия передней стенки живота (эмбриональная, пупочная грыжа, диастаз прямых мышц живота).
- Висцеромегалия (нефромегалия, гепатомегалия, спленомегалия).
- Макроглоссия.
- Необычное лицо (гипоплазия средней трети, гемангиома кожи лба, «насечки» на мочке ушной раковины).
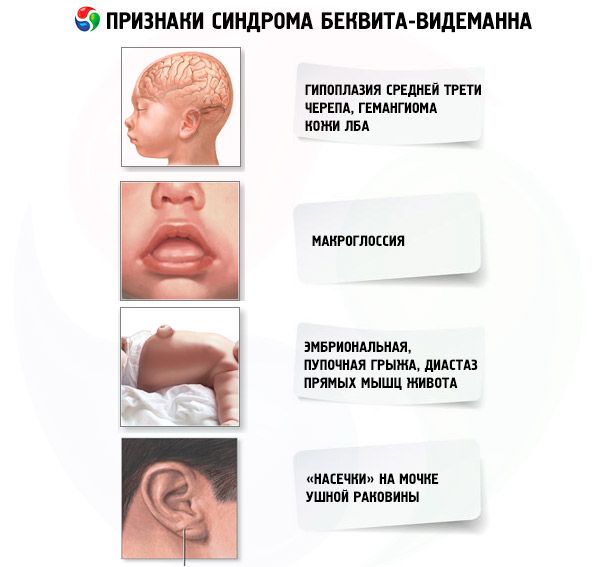
[22], [23], [24], [25], [26]
Какие анализы необходимы?
Лечение синдрома Беквита-Видеманна
Дефекты брюшной стенки устраняются с помощью хирургического лечения.
Гипогликемия у новорожденных с BWS должна лечиться в соответствии со стандартными протоколами терапии неонатальной гипогликемии.
Макроглоссия с возрастом часто становится менее заметной и не требует какого-либо лечения. В тяжелых случаях, макроглоссия устраняется с помощью операции. Некоторые хирурги рекомендуют выполнять оперативное вмешательство между 3 и 6 месяцами.
Гемигипертрофия в тяжелых случаях корректируется ортопедическими методами.
[27]
Прогноз
Синдром Беквита-Видеманна имеет разный прогноз для жизни. Он определяется своевременной диагностикой гипогликемии (профилактика умственной отсталости) и ранней диагностикой эмбриональных опухолей.
Продолжительность жизни, как правило, не отличается от здоровых людей.
Источник
Расскажу Вам свою историю , возможно кто-то в будущем по тегам #омфалоцеле или #синдром Беквита-Видемана , #СБВ #BWS найдет эту статью и меня для оказания моральной поддержки
В 2011 году я вышла замуж и через пол года мы решили планировать ребенка . Забеременела сразу в первый месяц , на втором узи сказали что будет мальчик ,все были в диком восторге . Но в то же время на узи нам сказали показаться в перинатальный центр , т.к. с размерами носика что-то не то . Пока я записалась и планировала туда попасть на прием у меня ребенок умер внутриутробно . Признаков абсолютно никаких небыло , ни крови ,ни выделений . Просто мои вспыхнувшие волнения по поводу отсутствия шевелений . 19 неделя беременности …
Итог : Больница , 2 дня капельниц вызывающих роды с последующим выскабливанием под наркозом .
2 Беременность наступила сразу как разрешили планировать (через пол года после случившегося ) на 7 неделе кровь -больница- выкидыш .
3 Беременность наступила не сразу , после кучи обследований и лечений , целый год мы ждали // . Но всё никак . Врач сказал даю 1-2 месяца если беременность не наступит будем делать стимуляцию. Мы уехали на отдых в Египет … 2014 год август .. оттуда берем путевку в Израиль по святым местам. Вифлеем, Храм Гроба Господня ,купание в реке Иордан . Нам провели какой-то индивидуальный ритуал в Храме Гроба Господня .
По приезду оттуда через неделю делаю тест и // . Счастью не было предела ,радость ,уверенность в том что на этот раз этот ребенок дан нам Богом и всё будет отлично … Но на первом скрининге в 11 недель нам ставят диагноз омфалоцеле и направляют в Перинатальный Кулаково . Там еще повторные узи в 12-13 недель . Далее в 15 недель на консилиуме нам говорят о том что сделают нам операцию ,если нет генетических отклонений у ребенка .
Микромолекулярный анализ на генетику показал хороший результат . Я делала амниоцентез . Также 1 пробирку с водами муж отвозил в другую лабораторию именно на Синдром Беквита-Видемана -не подтвердился (и слава богу ) ! Это в 18 недель беременности .
Прям под Новый год нам сообщили результаты и мы были счастливы что не придется делать аборт . Это для меня было бы адом …на тот момент я бы не пережила наверное.
Крайне тяжелая беременность , многоводие, живот как на двойню , кучу всего в придачу говорили УЗИсты , каждое узи (а их было очень много ) с ручьем слез . В основном сопутсвующие омфалоцеле патологии несовместимые с жизнью ребенка . 85 % из того что говорили УЗИсты не подтвердилось при рождении ребенка .
30 апреля 2015 года у меня отошли воды , буквально залило всю спальню. Это ровно 37 недель . Делали экстренное кесарево . Закричала сразу малышка , операционная была забита врачами , детскими хирургами , акушерами . Далее обнаружили какие-то серьезные кисты на обоих яичниках ,вызвали в срочном порядке хирурга отличного . Помню он сказал : Неужели на УЗИ ни один врач не посмотрел на яичники ??? Еще 1 неделя и они бы лопнули ! Провели операцию на оба яичника , сделали резекцию , но главное сохранили!
Вечером ко мне в реанимацию пришли врачи хирурги и сообщили что провели операцию у моей дочи по поводу омфалоцеле (поместили петли кишечника в животик в один этап) , она чувствует себя хорошо, перенесла хорошо , но завтра придет генетик , т.к. у них подозрения на Синдром Беквита-Видемана так как у ребенка увеличен язык .
Этот Синдром бывает у деток которые родились с диагнозом омфалоцеле ! Но не у всех , где-то у 20% детей с омфалоцеле может с последствии обнаружится этот синдром . Синдром достаточно хитрый (невыявляемый) , т.е. анализы могут его не показать !!!
У нашей дочери в 6 месяцев проведенный анализ крови на этот Синдром данный диагноз не подтвердил ! Но все внешние признаки говорят о 100% Беквите .
В 5,5 месяцев дочке была проведена операция в челюстно-лицевой хирургии на резекцию языка . Ей хирургически его уменьшили .
На умственное развитие этот Синдром никак не влияет (за исключением гипогликемии при рождении ) , поэтому она у нас абсолютно умненькая ,здоровенькая, хитренькая девчонка. Ей скоро 3 года. Она недавно начала болтать . Она всеми обожаемый ребенок в семье ! Родители ,бабушки ,дедушки , дяди, тети в ней души не чают !
Уверена что она спокойно будет учится в самой обычной школе , институте .
Единственное что меня волнует сейчас – это высокий риск развития рака почек ,печени до 8 лет . Мы постоянно на контроле у узистов .
Единственное, что меня будет волновать в будущем – это её дети , мои внуки и внучки. У них уже риск рождения с Синдромом будет 1 / 2 . (50% того что может родится ребенок с Синдромом как у мамы) Если у нас с мужем этот случай был случайностью ,т.е. у нас в роду ни у кого такого не было ,то у неё может быть случай более сложный . Формы бывают разные …
Я состою в группе “Омфалоцеле и гастрошиз ” в Вконтакте . Некоторые активные пользователи этой группы знают меня и скидывают мне контакты девочек у которых рождаются детки с Беквитом . Я знаю как это важно именно на начальном этапе оказать поддержку таким мамам , показать фотки моей дочери сейчас , сказать что это не конец жизни мамы , это не муки и прочее . У нас счастье ,у нас прекрасная ЗДОРОВАЯ дочь!
В свое время в роддоме я 3 дня ревела ,не могла представить КАК Я СПРАВЛЮСЬ ! ЗА ЧТО ЭТО МНЕ И прочее …. Это конечно был полный эгоизм с моей стороны . Легче мне стало как раз после того как я нашла девушку через -через у которой дочка была с таким диагнозом . Она расставила все по полкам ,я увидела не страшные фото в интернете , а эту прелесную девочку -реальный случай ,случайный случай у здоровых мамы и папы .
Но это жизнь , Господь дает нам испытания не просто так .
Сейчас у меня 4 ая беременность , тоже долгожданная , мы более 1 года как планируем . Пока срок 10 недель . Жду 1 скрининг , надеюсь только на лучшее !!!!!
Продолжение следует…..
Р. S : к сожалению не очень хорошее продолжение у меня. 2 скрининга прошли замечательно, без паталогий, всё отлично и на 20 неделе сердце малыша остановилось. На этот раз вызывали роды, самые настоящие схватки и я родила мертвого ребёнка. Это самое страшное видеть это… Впервые было со мной. В первую беременность открытия не было и ребёнка сняли под наркозом, я не видела ничего.
Источник