
Дети с синдромом раннего старения


Прогерия — синдром преждевременного старения, проявляющийся характерными изменениями кожи и внутренних органов. Это редкая генетическая аномалия, выявляемая у 1 человека из 4 миллионов. В мире насчитывается не более восьмидесяти наблюдавшихся случаев такой болезни. Этиопатогенетические факторы прогерии до конца не изучены.
Выделяют две морфологические формы патологии:
- Детская прогерия – синдром Хатчинсона — Гилфорда,
- Прогерия взрослых – синдром Вернера.
Термин «прогерия» в переводе с древнегреческого языка означает «раннее старение». Противоестественное истощение всех систем жизнеобеспечения обусловлено генетическим сбоем. При этом процесс старения ускоряется в десятки раз.
При синдроме Хатчинсона — Гилфорда у детей с задержкой физического развития появляются признаки старения: облысение, морщины, специфическая внешность. Их организм сильно изменяется: нарушается структура кожи, отсутствуют вторичные половые признаки, отстают в развитии внутренние органы. Затем быстро развиваются старческие недуги: тугоухость, артрозо-артриты, атеросклероз, инсульт или инфаркт, деминерализация костей. Восьмилетний ребенок с таким заболеванием выглядит и чувствует себя на 80 лет. В психическом развитии больные дети остаются абсолютно адекватными. Их интеллектуальное развитие не страдает. Они редко доживают до 13 лет. Мальчики болеют прогерией несколько чаще, чем девочки.

пример развития ребенка с детской прогерией (синдромом Хатчинсона — Гилфорда) от 1 года до 12 лет
Синдром Вернера обычно начинает проявляться клинически у молодых людей в возрасте 16-20 лет. Прогерия взрослых – ускоренное старение с поражением всех систем и высоким риском развития рака различной локализации. Геномная нестабильность, управляющая обычным процессом старения, приводит к целому ряду патологических изменений. Такие больные умирают к 30-40 годам, имея все симптомы глубокой старости.

пациентка со взрослой прогерией (синдромом Вернера) – до дебюта заболевания в 15 лет и с развитой формой в 48 лет
Прогерия — неизлечимое заболевание, «забирающее» детство у больных ребят и «превращающее» их в настоящих стариков. Регулярная и адекватная медицинская помощь позволяет замедлить необратимый процесс старения и уменьшить выраженность клинических симптомов. Для этого применяют лекарственные препараты, пищевые добавки, хирургические и физиотерапевтические методики.
Этиология
Основная причина прогерии — одиночная генетическая мутация, механизм образования которой в настоящее время остается неизвестным. Одни ученые считают, что истинная причина мутации кроется в наследственности родителей, другие – в воздействии на зародыш излучения во время проведения рентгенографии беременной женщине.
При синдроме Вернера нарушается процесс воспроизведения молекул ДНК, а при синдроме Хатчинсона-Гилфорда – биосинтез белка, определяющего форму ядер клеток. Генетические нарушения делают клетки неустойчивыми, что приводит к запуску непредвиденных механизмов старения. Большое количество протеина скапливается в клетках, которые перестают делиться. При этом оболочка ядра становится нестабильной, а клетки организма приходят в негодность и отмирают преждевременно. Мутация приводит к выработке усеченного белка прогерина, который является неустойчивым и быстро деградирует в пределах клетки. В отличии от цельного белка он не внедряется в ядерную пластину, которая находится под ядерной мембраной и участвует в организации хроматина. Ядерная подложка разрушается, что заканчивается серьезными проблемами. Прогерин накапливается в клетках гладкой мускулатуры сосудистой стенки. Дегенерация этих клеток — одно из ведущих проявлений болезни.
Прогерия у взрослых наследуется по аутосомно-рецессивному принципу. У детей мутация не передается по наследству, а возникает непосредственно в организме больного. Это не удивительно, поскольку носители погибают до репродуктивного возраста.
Негенетические факторы, оказывающие влияние на развитие заболевания:
- образ жизни,
- сопутствующие заболевания,
- климат,
- питание,
- состояние окружающей среды,
- переизбыток солнечного воздействия,
- курение,
- гиповитаминозы,
- психоэмоциональные факторы.
Симптоматика
У детей (синдром Хатчинсона — Гилфорда)
При рождении больной ребенок кажется обычным малышом. Клинические признаки прогерии появляются уже на первом году жизни. Некоторые дети до 2-3 лет развиваются правильно, а потом начинаются отставать от сверстников по росто-весовым показателям. Дети с прогерией имеют специфическую внешность, поскольку признаки заболевания характерны и уникальны. Все больные поразительно похожи друг на друга.

типичные дети с синдромом Хатчинсона — Гилфорда из разных семей)

4-летний мальчик с менее типичной формой синдрома Хатчинсона — Гилфорда
- Больные дети имеют диспропорциональный череп с крупной мозговой частью и мелкой лицевой. Их нос напоминает клюв птицы: он тонкий и заостренный. Нижняя челюсть слабо развита, подбородок мелкий, губы тонкие, уши оттопырены, глаза неестественно большие. Зубы растут в два ряда, они дефломированы и рано начинают выпадать. Именно этот набор специфических черт делает больных детей похожими на стариков.
- Скелетные аномалии — основной симптом патологии. Больные дети отличаются низким ростом, недоразвитыми ключицами и бедрами. Кости больных очень хрупкие, они часто ломаются, подвижность суставов ограничена. Часто отмечаются вывихи бедра. Проявлением болезни становится карликовость. Наблюдаются дефекты скелета и ногтей. Ногти желтые и выпуклые, напоминающие «часовые стекла». Больные дети поздно начинают сидеть и ходить, у них изменяется осанка. Некоторые не в состоянии передвигаться без посторонней помощи.
- Кожа и подкожно-жировая клетчатка становятся тонкими. Раннее старение у больных проявляется по-разному: кожный покров покрывается морщинами, его тургор снижается, веки отекают, уголки рта опускаются. Сухость и сморщенность кожи особенно сильно заметна на лице и конечностях. Волосы на голове выпадают, становятся редкими и пушковыми, отсутствуют ресницы и брови. Сквозь истонченную кожу на голове видна венозная сетка. Из-за отсутствия подкожного жира ребенок внешне напоминает скелет, обтянутый кожей. Сухая и морщинистая кожа местами атрофируется, на ней появляются крупные участки гиперпигментации, уплотнения и ороговения.
- Прочие симптомы: инфантилизм, пронзительный голос, мышечная гипотрофия, короткие руки, узкая и выпяченная грудная клетка.
У взрослых (синдром Вернера)
Первые клинические признаки синдрома Вернера появляются к 14-18 годам. До полового созревания пациенты развиваются нормально. Затем они начинают отставать в физическом развитии от сверстников, лысеют, седеют. Их кожа истончается, морщинится и приобретает нездоровую бледность. Руки и ноги выглядят очень худыми из-за атрофии подкожного жира и мышц.

После 30 лет в организме больных развиваются следующие патологические процессы:
- катаракта обоих глаз,
- охриплость голоса,
- мозоли на стопах,
- язвенно-некротические процессы в коже,
- дисфункция потовых и сальных желез,
- нарушение работы сердца,
- остеопороз, метастатическая кальцификация мягких тканей, остеомиелит,
- эрозивные остеоартриты,
- “склеродермическая маска” на лице,
- низкий рост, плотное и короткое тело, худые и сухие конечности,
- снижение интеллекта,
- деформация ногтей,
- появление крупных пигментных пятен на коже,
- горб на спине,
- экзофтальм при дисфункции щитовидной железы,
- лунообразное лицо при дисфункции гипофиза,
- атрофия яичек у мужчин, нарушение менструального цикла у женщин, ранний климакс.
Эпидермис кожи уплощается, соединительнотканные волокна склерозируются, подкожная клетчатка атрофируется и частично замещается соединительной тканью. Ограничение пассивных движений в суставах рук и ног проявляется невозможностью полного сгибания и разгибания конечности. Это обусловлено рубцовым стягиванием сухожилий и болевым синдромом.
К 40 годам у больных развиваются старческие хвори: появляются проблемы с сердцем, сахарный диабет, частые переломы рук и ног, боли в суставах, доброкачественные и злокачественные опухоли кожи, дисфункция паращитовидных желез. Онкообразования, инфаркт и инсульт, внутренние кровоизлияния — основные причины летального исхода при прогерии.
Симптомы патологии лишь напоминают процесс нормального старения. Признаки старения при прогерии имеют различную степень выраженности или появляются в ином порядке. При естественном старении рост ногтей замедляется, а при прогерии – останавливается полностью. У пожилых людей брови истончаются после выпадения волос на голове, а у больных прогерией – наоборот.
Диагностика

синдром Хатчинсона — Гилфорда

синдром Вернера
Диагностика прогерии не требует проведения специфических методик и исследований. Внешние признаки болезни настолько красноречивы, что диагноз ставят, опираясь лишь на симптоматику и данные визуального осмотра. Специалисты изучают персональный и семейный анамнез.
Дополнительные исследования показаны с целью выявления сопутствующих заболеваний. Больным назначают общий анализ крови, ее биохимическое исследование, рентгенографию костно-суставного аппарата, гистологическое исследование кожи, медико-генетическое консультирование.
Лечение
В настоящее время панацеи от прогерии не существует. Все способы лечения, которые когда-либо применялись, оказались неэффективными. Врачи с помощью современных методов пытаются остановить болезнь и не позволить ей усугубиться. Пациентов совместно лечат специалисты в области эндокринологии, терапии, кардиологии.

Чтобы облегчить состояние больных, врачи назначают:
- «Аспирин» с целью профилактики острой сердечной и сосудистой недостаточности – инфаркта и инсульта.
- Статины для снижения уровня холестерина в крови и профилактики атеросклероза – «Липостат», «Холетар», «Липтонорм».
- Антикоагулянты для предотвращения или замедления процесса тромбообразования – «Варфарекс», «Синкумарин».
- Препараты, содержащие гормон роста – «Гетропин», «Неотропин», «Динатроп». Они позволяют корректировать отставание в физическом развитии.
- Препараты, заживляющие раны и стимулирующие кровообращение при образовании язв – «Мефанат», «Бепантен».
- Гипогликемические лекарства при сахарном диабете – «Диабетон», «Манинил», «Глиформин».
Физиотерапевтические процедуры проводят с целью воздействия на жесткие и тугоподвижные суставы. Больным назначают электрофорез, рефлексотерапию, ЛФК, инфракрасные лучи, водные процедуры, грязелечение, УВЧ-терапию, магнитотерапию. Больным прогерией показано правильное питание, обогащенное витаминами и микроэлементами, умеренная физическая нагрузка, длительные прогулки на свежем воздухе, полноценный отдых.
Грудничков кормят через зонд специальными молочными смесями, содержащими добавки для набора массы тела. Молочные зубы удаляют, освобождая место постоянным зубам, которые у больных детей быстро прорезываются. Специалисты следят за состоянием сердечно-сосудистой системы, что позволяет вовремя выявить начинающиеся недуги. Хирургическое лечение также показано больным с синдромом раннего старения. С помощью ангиопластики или аортокоронарного шунтирования восстанавливают проходимость кровеносных сосудов.

Прогерия — неизлечимая патология, развитие которой остановить невозможно. Экспериментальное лечение взрослых с помощью стволовых клеток и ингибиторов фарнезилтрансферазы позволяет восстановить подкожно-жировую прослойку, общий вес, уменьшить ломкость костей. Прогноз заболевания всегда неблагоприятный. Больные умирают от острой коронарной недостаточности или онкопатологии. Профилактика прогерии невозможна из-за того, что заболевание носит генетический характер. Пожизненная терапия способна лишь облегчить и продлить жизнь больным. Непрерывный уход, кардиологическая помощь и физиотерапия — основные направления в терапии недуга.
Видео: примеры людей с синдромом преждевременного старения
Видео: ТВ-шоу о людях с прогерией
Источник
Общая информация
Прогерия, или синдром Хатчинсона-Гилфорда или синдром преждевременного старения, является врожденным, редким, смертельным генетическим заболеванием с поразительными чертами, напоминающими преждевременное старение.
Дети с прогерией обычно имеют нормальный вид в раннем детстве. В возрасте примерно от 9 до 24 месяцев у детей, подверженных заболеванию, начинаются глубокие задержки роста, что приводит к низкому росту и низкому весу. У них также появляются изменяется черты лица, характеризующиеся:
- непропорционально маленьким лицом по сравнению с головой;
- недоразвитая челюсть (микрогнатия);
- пороки развития и скученность зубов;
- выразительные глаза;
- маленький носик;
- тонкая синева вокруг рта.
Кроме того, ко второму году жизни у людей с синдромом преждевременного старения волосы на голове, брови и ресницы выпадают (алопеция), и волосы на голове могут быть заметно маленькими, белыми или светлыми. Дополнительные характерные признаки включают генерализованный атеросклероз, сердечно-сосудистые заболевания и инсульт, вывихи бедра, необычно заметные вены кожи головы, потерю слоя жира под кожей (подкожная жировая ткань), дефекты ногтей, скованность суставов, дефекты скелета и/или другие нарушения.
Согласно сообщениям в медицинской литературе, у людей с синдромом Хатчинсона-Гилфорда развивается преждевременное, широко распространенное утолщение и потеря эластичности стенок артерий (атеросклероз), что приводит к опасным для жизни осложнениям в детстве, подростковом или раннем взрослом возрасте.
Дети с прогерией умирают от болезней сердца (атеросклероза) в среднем в возрасте 14 лет с диапазоном от 8 до 21 года. Как и с любым человеком, страдающим сердечно-сосудистыми заболеваниями,
Синдром преждевременного старения вызван мутацией гена LMNA или ламина A. Белок ламина A представляет собой каркас, удерживающий вмести ядро клетки. Ученные считают, что дефектный белок ламина А, делает ядро нестабильным. Эта клеточная нестабильность, по-видимому, приводит к процессу преждевременного старения при прогерии.
Признаки и симптомы прогерии

У новорожденных с синдромом Хатчинсона-Гилфорда могут быть некоторые подозрительные признаки, присутствующие при рождении, такие как:
- необычно тугая, блестящая, твердая (т.е., «склеродермоподобная») кожа над ягодицами, верхними ногами и нижней частью живота;
- синеватое изменение цвета кожи и слизистых оболочек в средней части лица (срединно-лицевой цианоз);
- «скульптурный» нос.
Глубокая прогрессирующая задержка роста обычно проявляется примерно к 24 месяцам, что приводит к низкому росту и весу.
Согласно сообщениям в медицинской литературе, дети в возрасте 10 лет, как правило, имеют рост, близкий к росту среднего трехлетнего ребенка.
Ко второму году жизни также наблюдается недоразвитие (гипоплазия) костей лица и нижней челюсти (микрогнатия). Лицо кажется непропорционально маленьким по сравнению с головой, а кости спереди и по бокам черепа необычно заметны (лобное и теменное выпуклость).
У больных детей обычно есть дополнительные характерные черты лица, в том числе:
- маленький, тонкий, потенциально заостренный нос;
- необычно заметные глаза;
- маленькие уши с отсутствующими долями;
- тонкие губы.
Также могут присутствовать зубные аномалии, такие как:
- отсроченное прорезывание первичных (молочных) и вторичных (постоянных) зубов;
- неправильно сформированные, маленькие, обесцвеченные зубы;
- необычно повышенная частота кариеса.
Кроме того, ненормальная малость челюсти может привести к скученности зубов.
У детей с синдромом преждевременного старения волосы на голове обычно теряются примерно к двум годам. Волосы на голове могут быть тонкими, белыми или светлыми, в некоторых случаях такими могут сохраняться на протяжении всей жизни. Кроме того, брови и ресницы также могут быть потеряны в раннем детстве.
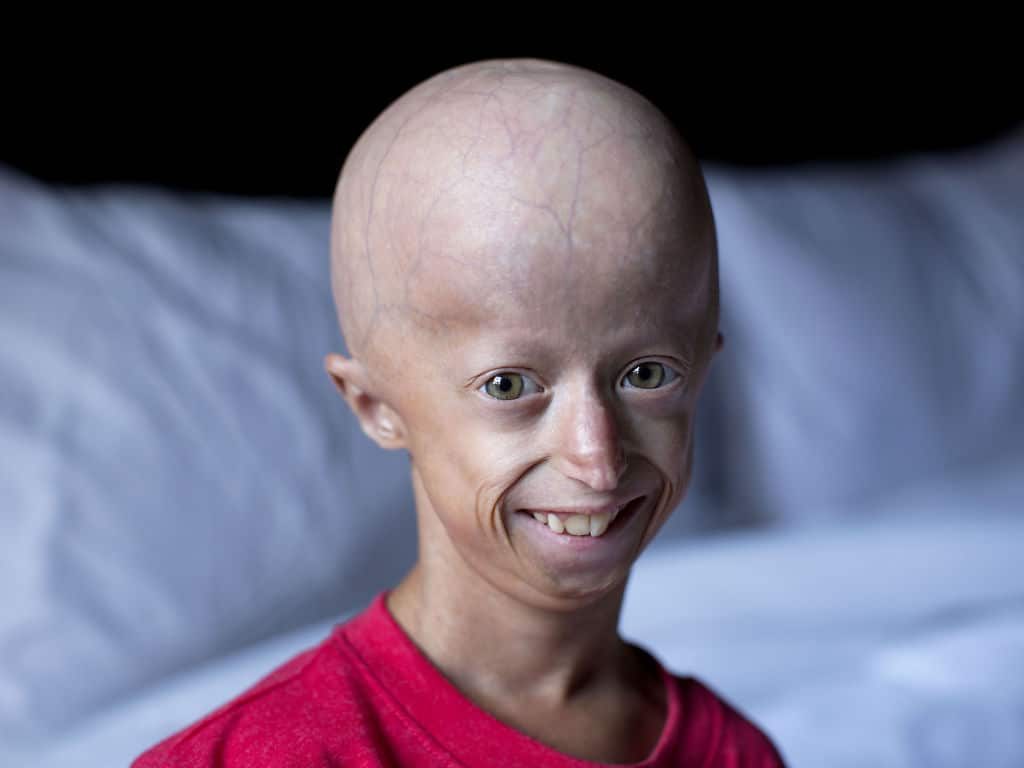
Синдром Хатчинсона-Гилфорда также характеризуется выраженными кожными нарушениями. Как обсуждалось выше, у новорожденных с таким расстройством могут быть «склеродермоподобные» изменения кожи на ягодицах, бедрах и нижней части живота. Кроме того, начиная с младенчества, происходит постепенная, почти полная потеря подкожной жировой ткани, и вены в определенных областях тела, особенно над кожей головы и бедрами, становятся ненормально заметными.
У больных детей кожа приобретает аномально состарившийся вид с необычно тонкими, сухими, морщинистыми и/или необычно блестящими и подтянутыми участками. Кроме того, с возрастом на участках кожи, подверженных воздействию солнца, могут появляться коричневатые кожные пятна. Больные дети обычно имеют дефекты ногтей, например, желтоватые, тонкие, хрупкие, изогнутые ногти или их полное отсутствие.
Дети с прогерией также имеют характерные дефекты скелета. К ним может относиться запоздалое закрытие «мягкого пятна» в передней части черепа (передняя родничок), аномально тонкой «куполообразной» части черепа и/или отсутствие определенных заполненных воздухом полостей внутри черепа, открывающийся в нос (околоносовые или лобные пазухи). Во многих случаях у детей также имеются:
- короткие, тонкие ключицы;
- узкие плечи;
- тонкие ребра;
- узкая или «грушевидная» грудь с выступающим животом.
Кроме того, длинные кости рук и ног могут казаться необычайно тонкими и хрупкими и склонны к переломам, особенно кости верхних рук (плечевые кости).
У многих детей с нарушениями скелета возникают дегенеративные изменения (остеолиз), которые могут поражать:
- ключицы;
- кости на концах пальцев (терминальные фаланги), в результате чего пальцы выглядят необычно короткими и «конусообразными»;
- тазобедренный сустав (вертлужная впадина).
Дегенеративные изменения в тазобедренном суставе могут привести к деформации тазобедренного сустава, при которой наблюдается аномальное увеличение угла кости бедра, боль в бедре и вывих бедра.
Кроме того, у многих больных детей вокруг определенных суставов (кисти, ступни, колени, локти и позвоночник) постепенно образуются аномальные фиброзные ткани (периартикулярный фиброз), вызывая необычную выпуклость, жесткость и ограниченное движение пораженных суставов.
Из-за скованности коленей, прогрессирующей деформации бедра, и другими связанными с мышечно-скелетными нарушениями дети с расстройством обычно имеют характерную, широко распространенную «стойку всадника» и шаркающую манеру ходьбы. Прогерия также связана с общей потерей плотности кости (остеопорозом), состоянием, вызывающим или способствующий повторным переломам после незначительной травмы.
Дополнительные симптомы и признака связанные с синдромом Хатчинсона-Гилфорда могут включать:
- сильно выделяющийся высокий голос;
- отсутствие груди или соска;
- отсутствие полового созревания;
- нарушение слуха и и прочие нарушения.
Согласно сообщениям в медицинской литературе, у пораженных детей в возрасте до 5 лет может развиться широко распространенное утолщение и потеря эластичности стенок артерий (атеросклероз). Такие изменения могут быть наиболее очевидными в определенных кровеносных сосудах, например, артериях, транспортирующих богатую кислородом кровь к сердечной мышце (коронарные артерии) и главной артерии тела (аорта).
Дополнительные признаки могут включать увеличение сердца (кардиомегалия) и аномальные шумы в сердце. В детском или подростковом возрасте прогрессирующий артериосклероз может привести к:
- периодическим болям в груди из-за недостаточного снабжения сердечной мышцей кислородом;
- затруднению кровотока в кровеносных сосудах головного мозга (цереброваскулярная окклюзия);
- прогрессирующей неспособности сердца эффективно перекачивать кровь в легкие и остальную часть тела (сердечная недостаточность);
- локализованной гибели части сердечной мышцы, вызванной прекращением ее кровоснабжения (инфаркт миокарда или сердечный приступ).
Причины прогерии
Ученные установили, что причиной возникновения прогерии является однобуквенная ошибка в гене на хромосоме 1, которая кодирует ламин A, белок, являющийся ключевым компонентом мембраны, окружающей ядро клетки. Аномальный белок ламина А, продуцируемый при синдроме, называется прогерин.
Прогерия обычно не передается по наследству. Изменения в генах — почти всегда случайное явление, встречающиеся крайне редко. Дети с другими типами прогероидных синдромов, которые не болеют синдромом Хатчинсона-Гилфорда, могут иметь заболевания, передающиеся по наследству.
Тем не менее, прогерия вызвана спорадической аутосомно-доминантной мутацией — спорадической, потому что это новое изменение в гене, и доминантной, потому что для того, чтобы синдром возник, необходимо изменение только одной копии гена. Для родителей, у которых никогда не было ребенка с прогерией, шансы родить ребенка с болезнью составляют 1 к 4-8 миллионам. Но для родителей, у которых уже есть ребенок с прогерией, шансы повторного рождения ребенка с заболеванием, намного выше — около 2-3%. Это связано с состоянием, называемым мозаицизмом, когда родитель имеет генетическую мутацию болезни в небольшой части своих клеток, но сам не болеет.
Конкретная первопричина ускоренного старения, связанная с синдромом Хатчинсона-Гилфорда, пока не известна. Многие ученные предполагают, что ненормальный процесс старения происходит из-за кумулятивного повреждения клеток в результате продолжающихся химических (метаболических) процессов в клетках организма.
Согласно этой теории, во время химических реакций, в организме образуются определенные соединения, называемые свободными радикалами. Считается, что увеличивающееся накопление свободных радикалов в тканях организма в конечном итоге приводит к повреждению и нарушению функционирования клеток, что в конечном итоге и приводит к старению. Считается, что некоторые ферменты (известные как антиоксидантные) играют роль в сдерживании процесса старения, способствуя устранению вредных свободных радикалов.
Ферменты — это белки, вырабатываемые клетками, ускоряющие скорость химических реакций в организме. Некоторые ученные указывают, что снижение активности некоторых ферментов может играть роль в ускоренном старении у людей с синдромом Хатчинсона-Гилфорда.
В одном исследовании клетки кожи (фибробласты), полученные от людей с прогерией, сравнивали с клетками кожи людей без заболевания. В фибробластах пациентов с прогерией уровни активности некоторых первичных антиоксидантных ферментов (например, глутатионпероксидазы [GPx], каталазы [CAT]) были значительно ниже, чем уровни, присутствующие в здоровых фибробластах.
Исследования показали, что прогерин вырабатывается гораздо более низкими уровнями у здоровых людей, и обычно накапливается в коронарных артериях в течение жизни с возрастом. Это открытие подтверждает возможность того, что прогерин является фактором риска развития атеросклероза среди населения земли в целом, и заслуживает изучения как потенциальной новый веток, помогающей прогнозировать риск сердечно-сосудистых заболеваний.
Таким образом, ученные подтвердили связь между нормальным старением, болезнями сердца и прогерией, поэтому поиск лекарства поможет не только больным детям, но, возможно, и тем, кто страдает от сердечных приступов, инсультов и других связанных со старением состояний.
Затронутые группы населения
Прогерия — редкое заболевание, одинаково влияющая на мужчин и женщин, и все расы. Это расстройство было первоначально описано в медицинской литературе в 1886 г. (Джонатан Хатчинсон) и 1897 г. (Гастингс Гилфорд). Распространенность синдрома составляет приблизительно 1 на 18 миллионов, таким образом, в любой конкретный момент в мире приблизительно 350-400 детей живут с прогерией.
Диагностика
Прогерия обычно диагностируется в течение второго года жизни или позже, когда прогероидные признаки становятся заметными. Диагноз основывается на тщательной клинической оценке, характерных физических данных, тщательном анамнезе пациента и диагностическом генетическом тестировании.
В более редких случаях заболевание можно заподозрить при рождении на основании распознавания определенных подозрительных данных (например, «склеродермоподобной» кожи над ягодицами, бедрами, нижней частью живота; цианозом средней части лица; «скульптурным» носом).
Для подтверждения или выявления определенных скелетных аномалий, потенциально связанных с расстройством, могут проводиться специализированные тесты. Кроме того, для оценки сопутствующих сердечно-сосудистых нарушений и определения надлежащего ведения заболевания могут также проводиться тщательная оценка состояния сердца и постоянный мониторинг (например, клинические обследования, рентгенологические исследования, специализированные исследования сердца).
Лечение прогерии
В сентябре 2012 года результаты первого в истории клинического исследования лекарственных препаратов для детей с прогерией показали, что Лонафарниб, тип ингибитора фарнезилтрансферазы, первоначально разработанный для лечения рака, оказался эффективным для лечения прогерии. Каждый ребенок продемонстрировал одно из четырех улучшений:
- увеличение веса;
- улучшение слуха;
- улучшение структуры кости;
- повышение гибкости кровеносных сосудов.
Помимо Лонафарниба, который не одобрен еще управлением по санитарному надзору за качеством пищевых продуктов и медикаментов и, следовательно, доступен только для клинических испытаний, лечение синдрома преждевременного старения направлено на конкретные симптомы, проявляющие у каждого человека. Лечение расстройств может потребовать скоординированных усилий группы специалистов, которым может потребоваться систематическое и всестороннее планирование лечения больного ребенка. Такими специалистами могут быть:
- педиатры;
- врачи, диагностирующие и лечащие нарушения скелета, мышц, суставов и других родственных тканей (ортопеды);
- врачи, диагностирующие и лечащие аномалии сердца и кровеносных сосудов (кардиологи);
- физиотерапевты и другие специалисты здравоохранения.
Специальные методы лечения для людей с синдромом являются симптоматическими и поддерживающими. Например, у пациентов с приступами боли в груди из-за недостаточного поступления кислорода в сердечную мышцу (ангинальные приступы) лечение может включать использование определенных лекарств, помогающих минимизировать такие симптомы.
Прогноз
Средняя продолжительность жизни людей с прогерией составляет 14 лет, хотя некоторые люди живут в возрасте старше 20 лет. Прогерия — это смертельный синдром.
Люди с заболеванием подвергаются повышенному риску многих заболеваний (вывихи, переломы, заболевания сердца, инсульт и проч.). У детей с прогерией очень часто развивается атеросклероз и суженные артерий. Большинство больных детей в конечном итоге умирают от болезней сердца.
Источник