
Фенилкетонурия врожденный гипотиреоз адреногенитальный синдром галактоземия и муковисцидоз


Неонатальный скрининг – обследование всех новорожденных детей на несколько наследственных заболеваний.
Цель: раннее выявление заболевания на доклинической стадии и организация лечения.
Рекомендации ВОЗ:
- Обследование проводится на заболевание ребенка, которое развивается постепенно и в манифестной фазе делает его инвалидом; при этом имеются проверенные методы предупреждения формирования патологического фенотипа;
- Тип наследования болезни и ее патогенез должны быть чѐтко установлены, а для семьи доступна медико-генетическая консультация;
- Методы скрининга, подтверждения диагноза и превентивного лечения должны быть доступны для практического здравоохранения;
- Ложно-положительные результаты методов скрининга должны быть редкими, ложно-отрицательные – исключены;
- Стоимость программ массового скрининга не должна превышать расходов на содержание и лечение детей, ставших инвалидами из-за данного заболевания (коэффициент “стоимость-эффективность” программ не должен превышать 1);
- Права семьи и самого ребенка, у которого по данным скрининга обнаружено наследственное (врожденное) заболевание, должны быть защищены (полная информация родителей о скрининг программе, право на отказ от включения их новорожденного в число обследуемых, конфиденциальность при подтверждении диагноза, сохранение врачебной тайны).
Обязательные элементы программы доклинической диагностики и массового скрининга:
– взятие биологического материала для исследования у новорожденных и доставка материала в диагностическую лабораторию (кровь из пяточки у ребенка должны взять на специальный тест-бланк на 4-е сутки жизни в роддоме, а в случае ранней выписки – на педиатрическом участке по месту жительства. У недоношенных детей анализ должны взять на 7-е сутки жизни; если роды произойдут вне родильного дома, необходимо без промедления обратиться в детскую поликлинику по месту жительства для проведения скрининга на 4-е сутки жизни ребенка)
– первичная лабораторная скрининг-диагностика
– уточняющая диагностика всех случаев с положительными результатами скрининга
– лечение больных и их диспансеризация с контролем и ходом лечения
– медико-генетическое консультирование семьи
Лабораторные методы массового скрининга:
1) Микробиологический метод Гатри,
2) Хроматография на бумаге или другом носителе (селикагель и др.),
3) Флуорометрический метод (более чувствительный), основанный на образовании флуоресцирующего комплекса фенилаланина с лейцил-аланином
4) Тандемная масс-спектрометрия(MS/MS),
5) Газовая хроматография с тандемной масс- спектрометрией(GC/MS)
Молекулярно-генетические методы диагностики наследственных болезней:
1) полимеразная цепная реакция (РСR)
2) блот-гибридизация
3) флуоресцентная ДНК-гибридизация insitu (FISH-метод)
4) рекомбинантные ДНК
5) геномная гибридизация
6) генетические микрочип-технологии
2)Целиакия( глютеновая болезнь, глютеноваяэнтеропатия, нетропическая спру, болезнь Ги-Гертера-Гейбнера): Распространенность среди населения разных стран -0.02-0.33%. Убольных в 40-100 раз повышается риск развития гастроинтестинальной карциномы или лимфомы.
Хроническое генетически детерминированное заболевание непереносимостью глютена с развитием атрофии сл.об. тонкой кишки и связанного с ней синдрома мальабсорбции.
Химический состав белка злаков: альбумин, глобулин, проламин, глютенин. Пшеница – глиадин, Рожь- секалин, Ячмень – гордеин, Овес – авенин.
Патогенез: однозначного мнения до настоящего времени нет. Основные гипотезы: генетическая (доказана ассоциацией с HLADO2 ИHLADO8), ИММУНОЛОГИЧЕСКАЯ, Дипептидазная, Вирусная.
Ведущие симптомы: 1) Манифестация – через 1.5-2 месяца после введения злаков в питание (возможно – после ОРВИ, кишечной инфекции). Длительная (до 10 лет и более) самопроизвольная ремиссия. 2)Снижение аппетита, рвота, метеоризмы, увеличение живота. 3)Стул: полифекалия, частота 2-20 раз в сутки, кал зловонный, серого цвета, пенистый, с жирным блеском. У 110% – запоры, 17% – нормальный стул. 4) Истончение подкожно-жирового слоя, снижение мышечного тонуса, гипотрофия, дистрофия ногтей, волос.
Внекишечные симптомы: Низкорослость, Резистентная железо-, фолиево-, В12-дефицитная анемия, лейкоцитопения, тромбоцитопения, Геморрагический синдром, Нарушение обмена кальция (судороги, остеопороз, остеомаляция, мышечная гипотония, боли в суставах), Рецидивирующий афтозный стоматит, Задержка полового развития, Нарушение со стороны ЦНС (апатия, нарушение сна, утомляемость, аггрессивность).
Классификация: Формы:
1)Типичная – проявляется классическими симптомами мальабсорбции, 2) Атипичная – проявляется каким-либо одним симптомом (чаще внекишечным) при отсутствии прочих, 3) латентная (скрытая) – нет явных клинических проявлений, но повышен титр специфических антител.
Методы диагностики: клинические симптомы, лабораторная диагностика, эндоскопия, HLAтипирование, серологические тесты, биопсия.
Б/Х анализ крови при целиакии: общий белок снижен, альбумины снижен, холестерин снижен, натрий, калий, кальций, фосфор снижены, железо, магний, цинк снижены.
Б/Х анализ кала при целиакии: общие жирные кислоты повышены ++, свободные жирные кислоты +++, энтерокиназа- – .
Эндоскопические признаки: специфических эндоскопических признаков не существует. Отсутствие складок, кишка в виде трубки, поперечная исчерченность складок.
Чувствительность и специфичность определения в крови различных антител:
ЕМА (антитела к эндотелию соединительнотканной субстанции, окружающей гладкомышечные элементы собственной пластинки) – чувствительность 97% у детей значительно вальирует, специфичность 98%.
IgAAGA – чув-ть 52%, спец-ть81-94%
Ant-TG – чув-ть 90%?Ложноотрицателен у детей до 2 лет, 96% спец-ть.
HLAтипирование- (у пациентов с предрасположенным диагнозом, по сомнительным результатам гистологического исследования): HLADQ2 (95% больных), HLADQ8 (5%БОЛЬНЫХ), Необхоимо, но недостаточное условие для подтверждения цилиакии (обнаруживаются у 30 процентов здоровых лиц.)
Принцип терапии: 1)пОжизненнаябезглютеновая диета. 2)В периоде клинической манифестации – учет и коррекция водно-электролитных нарушений, степени гипотрофии, вторичная лактазная недостаточность, нарушение функции почек и сердечно-сосудистой системы. 3)В периоде ремиссии – лечение дефицитных состояний и сопутствующих соматических заболеваний: анемии, нарушение минерального обмена, дисбиотических нарушений кишечника.
Безглютеновые продукты: содержат глютена не более 20мг кг сухого вещества. Рис, гречиха, кукуруза. Картофель, соя, морковь, капуста, кабачок, тыква, яблоки, груши, бананы, ффруктовые соки, мясо птицы, рыба, маргарин, растительные масла, мед, варенье, джемы.
Этапная диета при целиакии:
Исключается пожизненно: мука, хлеб, выпечка, крупы и макаронные изделия из пшеницы, ржи, ячменя и овса. Исключается в период клинической манифестации: молоко, пресные смеси, бобовые, фрукты и овощи с губой клетчаткой, животные жиры, жилистые жирные сорта мяса. Ограничиваются в период клинической манифестации: кисломолочные продукты, творог, сметана, сыр, сливочное масло, сахар, фрукты, цельные фруктовые соки. Рекомендуются в период клинической манифестации: Смеси на основе гидролиза БКМ, соевые смеси, безглютеновые безмолочные каши, овощное, картофельное пюре, мясное, рыбное пюре, желток, растительное масло, печеное яблоко, бананы, соки, разбавленные водой. Рекомендуются в период стойкой ремиссии: безглютеновые заменители хлеба, крупы, мука и крахмалы, рисовые, кукурузные, гречневые, пшено, картофель, орехи, молоко и молочные продукты, все овощи и фрукты, яйца, мясные и рыбные блюда без муки, растительное и оливковое масло.
Продукты содержащие скрытый глютен: клбасы, сосиски, полуфабрикаты из измельченного мяса и рыбы, мясные и рыбные консервы, особенно в томатном соусе. Овощные и фруктовые консервы. Многие виды детских консервированных блюд, прикормка. Мороженое, йогурт, плавленный сыр, маргарин. Некоторые виды уксусов и салатных соусов, майонезов. Концентрированные сухие супы, бульонные кубики. Быстрорастворимый кофе, чай для быстрого приготовления. Многокомпонентные сухие приправы и пряности. Имитация морепродуктов – крабовые палочки. Карамель, соевые и шоколадные конфеты с начинкой, восточные сладости, повидло промышленного производства, пищевые добавки, квас, пиво.
Повторный осмотр, визиты: Всем пациентам нужно дать письменные инструкции о диете на протяжении всей жизни. У пациентов с хорошим эффектом от аглютеновой диеты – каждые 6-12 мес для оценки симтоматики, статуса питания и согласия продолжать диету, анализов крови. Важно осматривать пациентов в их стрессовых ситуациях (особенно во время беременности). ВОП должен знать о возможных осложнениях и направлять к специалисту при необходимости.
Что делать есои эффект от диеты слабый: убедиться что пациент соблюдает диету (или хочет ее собдюдать), подтверждение диагноза (институт гастроэнтерологии), исключить другие заболевания.
БИЛЕТ №6
Дата добавления: 2016-12-05; просмотров: 2251 | Нарушение авторских прав | Изречения для студентов
Читайте также:
Рекомендуемый контект:
Поиск на сайте:
© 2015-2020 lektsii.org – Контакты – Последнее добавление
Источник
15.09.2020

В отличие от врожденных заболеваний, наследственные болезни возникают в результате генетических нарушений. Иногда болезнь проявляет себя через некоторое время и при определенных факторах, которые являются пусковыми для ее развития. В иных случаях малыши уже рождаются больными.
Ранняя диагностика позволяет точно поставить диагноз, принять решение о сохранении беременности или подготовиться к длительному лечению ребенка.
Пренатальный скрининг наследственных болезней
Многие хромосомные и генные нарушения чреваты для плода врожденными патологиями, в том числе, несовместимыми с жизнью. Например, малыши с синдромами Эдвардса и Патау часто гибнут и не доживают до года. Никакие медицинские меры, лекарства и способы не помогают продлить их существование. Причина этих синдромов — трисомия, когда вместо положенных двух копий, в результате генетических нарушений, образуется три копии определенной пары хромосом. При синдроме Эдвардса утроение происходит по 18-й хромосоме, а при синдроме Патау по 13-й хромосоме.
А вот при синдроме Дауна (трисомия по 21-й хромосоме), который встречается довольно часто — один ребенок на 800 родов, выживаемость выше (в среднем такие люди живут до 36 лет). При своевременной регулярной психофизиологической коррекции и уходе, детки вполне социализируются и могут найти свое место в обществе.
Для диагностики синдромов Дауна, Патау, Эдвардса и числовых нарушений половых хромосом, которым в большей степени подвержен плод у женщины позднего репродуктивного возраста, в СИТИЛАБ проводят пренатальный биохимический скрининг, а также новые уникальные неинвазивные ДНК-тесты PrenaTest®.
4903 —Неинвазивный пренатальный тест PrenaTest® (трисомия по 21 хромосоме + определение пола) (с 9-й недели беременности).
4904 —Неинвазивный пренатальный тест PrenaTest® (трисомия по 21, 13, 18 хромосомам + определение пола (с 9-й недели беременности).
4905 —Неинвазивный пренатальный тест PrenaTest® на определение наличия у плода трисомии по 21, 13 и 18 хромосоме (синдромы Дауна, Патау, Эдвардса) + числовые нарушения половых хромосом и определение пола (с 9-й недели беременности).
99-20-301 — Пренатальный скрининг трисомий (1 триместр беременности: 11-13 недель + б дней).
97-20-323 — Пренатальный скрининг, I триместр (10-13 недель) — PRISCA I.
99-20-302 — Пренатальный скрининг трисомий (2 триместр беременности: 14-20 недель + 6 дней).
97-20-324 — Пренатальный скрининг, II триместр (14-20 неделя) — PRISCA II.
Неонатальный скрининг новорожденных
Данный вид скрининга является обязательным для всех новорожденных и необходим для выявления таких наследственных заболеваний, как фенилкетонурия, муковисцидоз, врожденный гипотиреоз, адреногенитальный синдром, галактоземия. Кровь для исследования берется у новорожденного на 4-7 день после рождения. После выявления нарушений (маркеры заболевания), родители получают направление к узкопрофильному специалисту для дальнейшей консультации.
Фенилкетонурия. Причина — нарушение обмена аминокислот, в первую очередь фенилаланина. Вызывает поражение центральной нервной системы, что приводит к умственной отсталости ребенка. В основе лечения — пожизненная низкобелковая диета с исключением ряда продуктов, содержащих аминокислоту фенилаланин (мясо, рыба, яйца, сыр и др.).
Муковисцидоз. Вызывает поражения органов дыхания, поджелудочной железы. Симптомы выявляются не сразу, а через некоторое время после рождения. Заболевание провоцирует хронические бронхиты, рецидивирующий панкреатит, дыхательную недостаточность и, как следствие, отставание в физическом развитии ребенка. Лечится симптоматически, важную роль также играет диета с достаточным количеством белка.
Врожденный гипотиреоз. Чаще регистрируется у девочек, проявляется сразу после рождения в виде полной или частичной дисфункции щитовидной железы. Это приводит к угнетению всех физиологических процессов в организме. Лечение начинается с момента рождения и продолжается всю жизнь (прием L-тироксина).
Галактоземия. Недостаточность ферментов, участвующих в метаболизме галактозы (простой сахар) приводит к патологиям ЖКТ, нервной системы детей раннего возраста. Предполагается, что такой ребенок пожизненно будет соблюдать довольно жесткую специальную диету.
Ранняя диагностика вышеперечисленных заболеваний позволяет выявить проблему в первые дни жизни малыша. Это позволяет своевременно начать необходимое лечение и программу профилактики, которые существенно улучшают качество жизни ребенка.
Источник

Неонатальный скрининг, ласково именуемый в нашей стране «пяточка», является одним из первых важных исследований новорожденного. В России скрининг преимущественно направлен на выявление пяти наследственных болезней обмена: фенилкетонурии, врожденного гипотиреоза, врожденной дисфункции коры надпочечников (ВДКН), галактоземии и муковисцидоза. За рубежом этот список расширен до 50 различных заболеваний, в некоторых штатах Америки их свыше 60. Здоровый доношенный новорожденный допускается к скринингу на 4–5 сутки, недоношенный — на седьмой день после рождения. Заболевания, на выявление которых направлен скрининг, никак не проявляют себя в периоде новорожденности, но их ранняя диагностика и своевременно начатое патогенетическое лечение существенно влияют на прогноз и качество жизни ребенка. Помимо исследования крови проводится аудиометрия для оценки слуха и пульсоксиметрия для скрининга пороков сердца, но в данной статье мы преимущественно сосредоточимся на тестировании крови.
Разработка программы скрининга началась в шестидесятых годах прошлого века, когда Роберт Гатри создал технологию тестирования сухих отпечатков крови на фильтровальной бумаге. Первым заболеванием, которое стало кандидатом для массовой диагностики, была фенилкетонурия, так как ее раннее выявление и коррекция питания способны предотвратить развитие тяжелых неврологических нарушений. Затем к скринингу добавилось еще несколько заболеваний: врожденный гипотиреоз, ВДКН, галактоземия и муковисцидоз. Тандемная масс-спектрометрия (ТМС) позволила значительно расширить список заболеваний, добавив к болезням обмена веществ гемоглобинопатии, спинальную мышечную атрофию, тяжелый комбинированный иммунодефицит и др.
Таблица 1 | Рекомендуемая The American College of Obstetricians and Gynecologists (ACOG) скрининг-панель для врожденных заболеваний
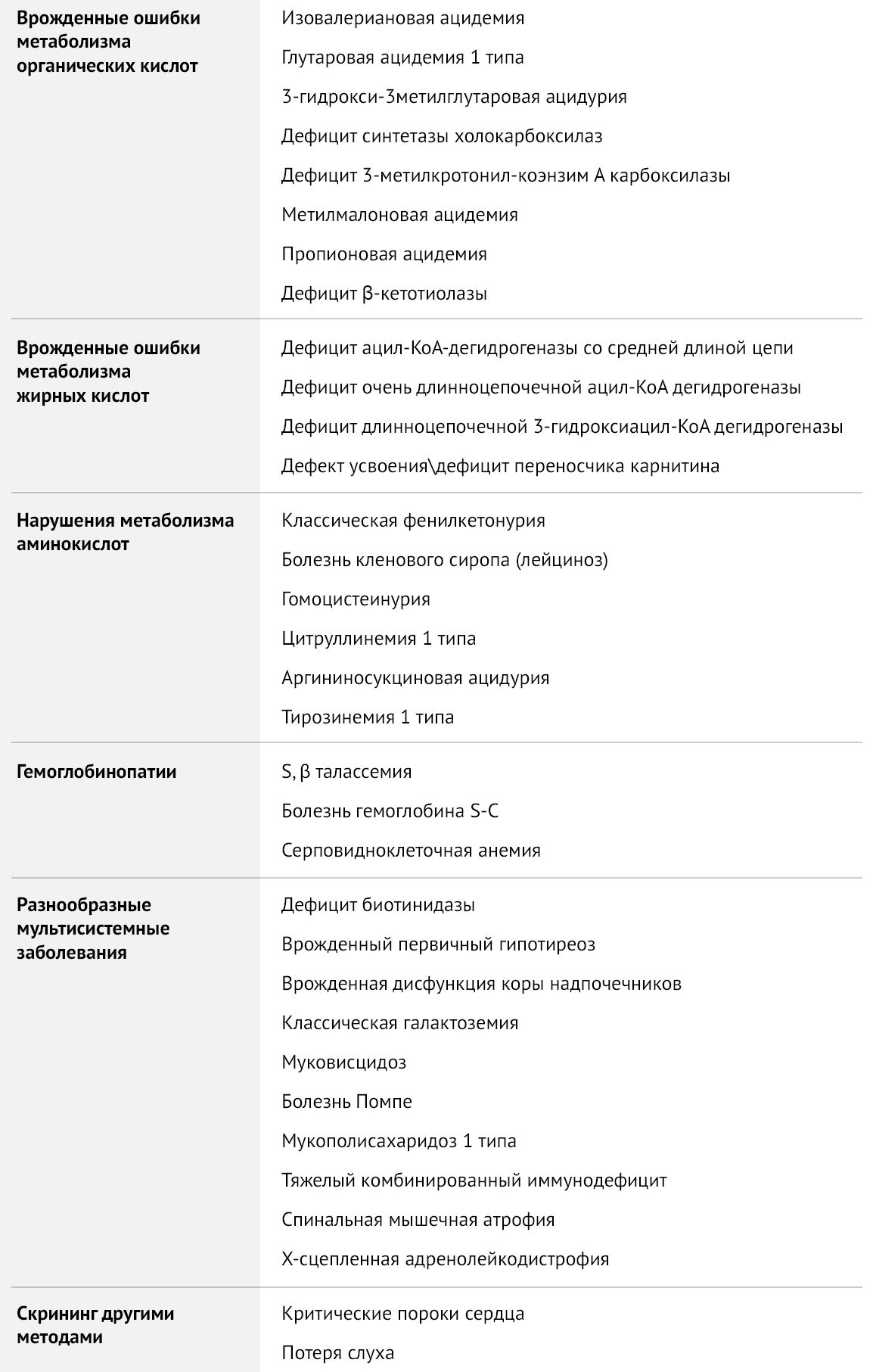
В данной статье будут рассмотрены два скрининга, доступные в нашей стране: обязательный, включающий тестирование на пять заболеваний (врожденный гипотиреоз, ВДКН, фенилкетонурия, галактоземия, муковисцидоз) и расширенный скрининг на наследственные нарушения метаболизма.
На 4–5 сутки после рождения здорового доношенного ребенка или на седьмые сутки жизни недоношенного ребенка проводится тестирование методом «сухого пятна».
Фенилкетонурия (в современной классификации ― ФАГ-зависимая ФКУ) обусловлена мутацией гена фенилаланингидроксилазы и относится к числу аминокислотных аминоацидопатий. В норме фенилаланин (ФА) путем реакций гидроксилирования превращается в тирозин, однако в случае мутации вышеназванного гена активность превращающего фермента снижается, создается дефицит тирозина одновременно с избытком ФА, образующего токсичные метаболиты (фенилацетат, фенилпируват, фениллактат). Снижение образования тирозина влечет за собой нарушение синтеза гормонов щитовидной железы, нейротрансмиттеров и пигментов меланоцитов, а избыток ФА приводит к дисбалансу аминокислот в тканях мозга, обусловленному торможением их всасывания в желудочно-кишечном тракте или нарушением реабсорбции из почечных канальцев, нарушению образования или стабилизации полирибосом, снижению синтеза миелина, норадреналина и серотонина. Также за счет конкурентного ингибирования создается дефицит тирозиназы, что в совокупности с дефицитом тирозина приводит к снижению образования меланина и гипопигментации.
Основной проблемой пациентов с ФКУ являются нарушения функции ЦНС: от сонливости, вялости, отсутствия аппетита в период манифестации в 2–6 месяцев до тяжелых нарушений психомоторного развития в будущем; нередко развиваются атаксия, гиперкинезы, тремор рук, парезы по центральному типу. Единственный способ предотвратить развитие вышеназванных нарушений — назначение гипофенилаланиновой диеты с момента рождения с поддержанием низкого уровня фенилаланина в течение всей жизни.
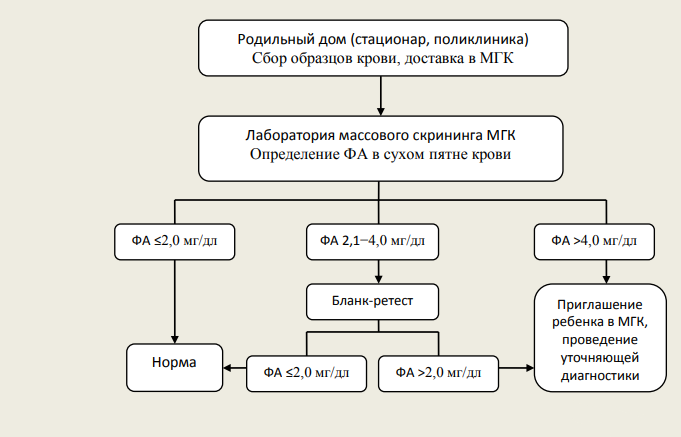
Рисунок 1 | Интерпретация результатов исследования на наличие фенилкетонурии
ВДКН обусловлена дефицитом ферментов и транспортных белков, участвующих в биосинтезе кортизола. Наиболее часто встречается дефицит 21-гидроксилазы, что в свою очередь приводит к дефициту кортизола и альдостерона и ответному увеличению секреции АКТГ и гиперплазии коры надпочечников. В условиях дефицита фермента происходит значительное накопление предшественников гормонов, что приводит к увеличению синтеза тестостерона, не зависящего от 21-гидроксилазы. В итоге у пациента формируется надпочечниковая недостаточность и гиперандрогения. Гормональным маркером дефицита 21-гидроксилазы является уровень 17-гидроксипрогестерона (17-ОНП), определяемый в рамках неонатального скрининга. Результат трактуется как положительный, если при двукратном тестировании образца уровень 17-ОНП у доношенных новорожденных составляет ≥ 20 нг/мл. У недоношенных детей при заборе крови на 7–8 сутки после рождения скрининговый результат трактуется как положительный при следующих уровнях 17-ОНП: на сроке 23–32 недели гестации ― ≥ 65 нг/мл; на сроке 33–36 недель гестации ― ≥ 40 нг/мл.
Врожденный гипотиреоз в большинстве случаев вызван дефектами самой щитовидной железы (первичный гипотиреоз). Причины первичного врожденного гипотиреоза можно в широком смысле классифицировать как неспособность щитовидной железы нормально развиваться (дисгенезия) или неспособность структурно нормальной щитовидной железы производить нормальные количества гормона (дисгормоногенез). Дисгенезия щитовидной железы, охватывающая весь спектр агенеза, гипоплазии и эктопии, является наиболее частой причиной врожденного гипотиреоза. В то время как это заболевание остается наиболее частой причиной врожденного гипотиреоза, частота возникновения дисгормоногенеза за последние несколько десятилетий увеличилась. В то время как на дисгормоногенез приходится только 15 % врожденного гипотиреоза, диагностированного в первые дни скрининга новорожденных, у 30–40 % младенцев, прошедших скрининг по современным протоколам, имеется эктопическая щитовидная железа, соответствующая одной из форм дисгормоногенеза. В отличие от дисгенезии щитовидной железы, при которой моногенная причина присутствует только у небольшого количества пациентов, дисгормоногенез часто возникает из-за генетического дефекта на каком-либо этапе синтеза тиреоидных гормонов.
Учитывая разнообразие функций тиреоидных гормонов в организме человека, врожденный гипотиреоз характеризуется разнообразием клинических проявлений с поражением всех органов и систем. При отсутствии своевременного лечения на первый план выходит задержка психомоторного и речевого развития, затем наступают отставание в физическом развитии и задержка полового развития. Основной задачей скрининга является наиболее раннее выявление детей с подозрением на врожденный гипотиреоз.

Рисунок 2 | Интерпретация результатов исследования на наличие врожденного гипотиреоза
Галактоземия — аутосомно-рецессивное наследственное нарушение обмена углеводов, при котором в организме накапливается избыток галактозы и ее метаболитов. В норме галактоза образуется в результате гидролиза лактозы в кишечнике либо в процессе ферментных реакций, обмена гликопротеинов и гликолипидов. Галактоза является материалом для образования клеточных мембран, нервной ткани, нервных окончаний и т. д. В результате ферментных реакций она превращается в глюкозу, и именно дефицит галактозо-1-фосфатуридилтрансферазы лежит в основе патогенеза данного заболевания. Метаболиты галактозы обладают повреждающим действием. Так, галактитол проникает в хрусталик глаза, приводя к повышению осмотического давления, электролитным нарушениям и денатурации белка с формированием катаракты. Другие метаболиты обладают гепато-, нейро- и нефротоксическим действиями, а также вызывают гемолиз эритроцитов. Тормозящее влияние метаболитов галактозы на углеводный обмен приводит к гипогликемии.

Рисунок 3 | Интерпретация результатов исследования на наличие галактоземии
Муковисцидоз — аутосомно-рецессивное заболевание, связанное с мутацией гена МВТР (трансмембранного регулятора муковисцидоза). МВТР является хлорным каналом, мутации гена которого нарушают не только транспорт, но и секрецию ионов хлора. При затруднении их прохождения через клеточную мембрану увеличивается реабсорбция натрия железистыми клетками, нарушается электрический потенциал просвета, что вызывает изменение электролитного состава и дегидратацию секрета желез внешней секреции. В результате выделяемый секрет становится чрезмерно густым и вязким. Поражаются все экзокринные железы организма: печень, поджелудочная железа, мочеполовая система, но наиболее ярко муковисцидоз проявляет себя со стороны органов дыхания, провоцируя бронхообструкцию, дыхательную и сердечную недостаточность, легочную гипертензию.
Рисунок 4 | Интерпретация результатов исследования на наличие муковисцидоза
Органические ацидемии — группа аутосомно-рецессивных наследственных заболеваний обмена, в основе патогенеза которых лежит дефицит ферментов, участвующих в метаболизме белков, что приводит к повышению уровня кетоновых тел, обладающих токсическим действием на различные органы и ткани, в частности, на ЦНС. Данные заболевания манифестируют уже в стадии декомпенсации, как правило, в период с первой недели до первого года жизни. Триггерами служат стресс, длительное голодание, инфекционные заболевания, иммунизация, реже — чрезмерное употребление белковой пищи. Проявляются преимущественно неврологической симптоматикой: нарушение сознания вплоть до комы, эпилептические приступы, нарушение мышечного тонуса, у детей старшего возраста — нарушения психоречевого развития, атаксия, очаговые неврологические симптомы, синдром Рейе (острая печеночная недостаточность, сочетающаяся с энцефалопатией), психические расстройства.
Нарушения окисления жирных кислот — врожденный дефект метаболизма из-за нарушения либо митохондриального β-окисления, либо транспорта жирных кислот с использованием карнитинового транспортного пути. Проявления зависят от нарушения метаболизма конкретной кислоты, но все они имеют общие черты и требуют схожей тактики лечения. В периоде новорожденности метаболические нарушения проявляются тяжелой кардиомиопатией, гипокетотической гипогликемией, дисфункцией печени в первые несколько дней или недель жизни, часто заканчиваясь летально. В младенческом и детском возрасте характерны эпизоды летаргии и рвоты, развивается дисфункция печени и гипокетотическая гипогликемия, энцефалопатия, что может привести к внезапной младенческой смерти. У подростков и во взрослом возрасте дебютируют эпизодическим рабдомиолизом, мышечной слабостью, миалгией. Лечение включает отказ от голодания, симптоматическую терапию развившихся осложнений и включение в рацион добавок, если это необходимо.
Болезнь кленового сиропа (она же лейциноз) — наследственное заболевание, обусловленное дефицитом дегидрогеназы кетокислот с разветвленной цепью и нарушением метаболизма лейцина, изолейцина, валина (аминокислоты с разветвленной цепью, АКЦР). Повышение уровня АКЦР и их метаболитов, в частности, кетокислот, приводит к кетоацидозу, атрофии ткани головного мозга, нарушению окислительного фосфорилирования в дыхательной цепи митохондрий. Избыток лейцина обладает нейротоксическим эффектом, вызывая дисфункцию астроцитов, апоптоз нейронов и блокируя транспорт через гематоэнцефалический барьер аминокислот, важных для синтеза нейротрансмиттеров.
Гомоцистеинурия — наследственное заболевание из группы аминоацидопатий, обусловленное нарушением метаболизма серосодержащих аминокислот, в частности, метионина. Дефицит цистатион-b-синтазы нарушает преобразование метионина в цистеин. Высокий уровень гомоцистеина связан с образованием некротически-дегенеративных участков в почках, селезенке, слизистой оболочке желудка и сосудах, активацией XII фактора свертывания, способствующего тромбообразованию.
Аргининосукциновая ацидурия вызывается мутациями в гене ASL, который кодирует фермент аргининосукцинатлиазу. Этот фермент катализирует превращение аргинино-янтарной кислоты в аргинин и фумарат на четвертом этапе цикла мочевины. Дефекты на этой стадии цикла мочевины приводят к накоплению в плазме аммиака, аргинино-янтарной кислоты, цитруллина и оротовой кислоты в моче, а также к дефициту аргинина в плазме. Ацидурия может иметь различную клиническую картину с началом в любом возрасте, включая период новорожденности. Состояние новорожденных обычно не вызывает подозрений в течение первых 24–48 часов после рождения, но в течение нескольких дней дебютирует тяжелая гипераммониемия, проявляющаяся летаргией, сонливостью, отказом от еды, рвотой, тахипноэ и респираторным алкалозом. Если не начать лечение, может произойти обострение летаргии, судороги, кома и смерть. Позднее начало ацидурии обычно индуцировано острой инфекцией, стрессом или высоким потреблением белка. Сообщалось также о поздних когнитивных дефектах или нарушениях обучаемости при отсутствии эпизодов гипераммониемии. У некоторых пациентов заболевание может протекать бессимптомно, несмотря на четкие биохимические признаки.
Тирозинемия 1 типа — заболевание, обусловленное дефицитом фумарилацетоацетатгидролазы, в результате чего происходит накопление высокотоксичных фумарил- и малеилацетоацетата, обладающих гепатотоксическим и канцерогенным действием. Конечные метаболиты — сукцинилацетон и сукцинилацетоацетат — являются митохондриальными токсинами, тормозящими фосфорилирование и блокирующими цикл Кребса. Накопление токсинов приводит к прогрессирующему заболеванию печени с развитием печеночной недостаточности, цирроза, тубулопатии с формированием ренальной тубулопатии, гипофосфатемического рахита, синдрома Фанкони. Острая тирозинемия сопровождается развитием гипертрофической кардиомиопатии. Кроме того, нарушается путь синтеза порфирина, ингибируется синтез порфобилиногена, что приводит к кризам, проявление которых напоминает порфирию. Все пациенты подвержены высокому риску развития гепатоцеллюлярной карциномы, вторичной по отношению к циррозу. Без своевременного лечения дети погибают в возрасте 10 лет.
- Rink B., Dukhovny S. Newborn Screening and the Role of the Obstetrician-Gynecologist //OBSTETRICS AND GYNECOLOGY. – 2019.
- Merritt J. L., II M. N., Kanungo S. Fatty acid oxidation disorders //Annals of translational medicine. – 2018.
- Еремина Е. Р. Клинический случай редкой органической ацидурии //Медицинская генетика. – 2018.
- Cherella C. E., Wassner A. J. Congenital hypothyroidism: insights into pathogenesis and treatment //International journal of pediatric endocrinology. – 2017.
- Чикулаева О. А. Федеральные клинические рекомендации по диагностике и лечению врожденного гипотиреоза у детей //Проблемы эндокринологии. – 2014.
- Клинические рекомендации “Фенилкетонурия и нарушения обмена тетрагидробиоптерина у детей”, 2017.
- Клинические рекомендации “Федеральные клинические рекомендации по оказанию медицинской помощи детям с галактоземией”, 2015
- Клинические рекомендации “Кистозный фиброз (муковисцидоз)”, 2020
- Клинические рекомендации “Гомоцистеинурия”, 2016
- Клинические рекомендации “Клинические рекомендации по ведению и терапии новорожденных с заболеваниями надпочечников”. 2016
- De Laet C. et al. Recommendations for the management of tyrosinaemia type 1 //Orphanet journal of rare diseases. – 2013.
- Nagamani S. C. S., Erez A., Lee B. Argininosuccinate lyase deficiency //Genetics in medicine. – 2019.
Источник