
Фрагильная х хромосома синдром мартина белла

Синдром Мартина-Белл – это наследственная болезнь, которая характеризуется стойким интеллектуальным снижением, расстройствами аутистического спектра и специфическими фенотипическими особенностями. Ключевой симптом – недостаточность познавательных функций. Отмечается гиперактивность, дефицит коммуникативных способностей, замкнутость. Лицо удлиненное, ушные раковины большие, лоб выступающий, кончик носа загнутый. Диагностика основывается на клинико-анамнестических данных и результатах биогенетического анализа. Лечение симптоматическое, включает использование медикаментов и психолого-педагогическую коррекцию.
Общие сведения
Синдром Мартина-Белл получил свое название по фамилиям исследователей, впервые описавших патологию. В 1943 году физиологи из Великобритании Д. Мартин и Д. Белл изучали 11 случаев олигофрении у мужчин из одной семьи, в которой женщины имели нормальное интеллектуальное развитие. Генетическая основа заболевания была выявлена в 1969 году американским генетиком Г. Лабсом. Синонимичное название – синдром ломкой X-хромосомы. Распространенность среди мальчиков составляет 1:4 000, среди девочек – 1:6 000. Согласно данным зарубежных врачей-генетиков, частота синдрома Мартина-Белл у пациентов мужского пола с умственной отсталостью достигает 1,9-5,9%. Отечественные исследования указывают на более высокие значения, в соответствии с ними этот синдром имеют 8-10% больных олигофренией.
Синдром Мартина-Белл
Причины
Синдром Мартина-Белл является результатом дефекта гена FMR1, расположенного в X-хромосоме. Наследование происходит по доминантному сцепленному с полом типу с неполной пенетрантностью. У мужчин присутствует одна X-хромосома, поэтому мутантный аллель всегда провоцирует болезнь. У женщин есть две половые хромосомы типа X: одна активная, другая – резервная, инактивированная. Таким образом, при наличии мутации в одном из двух генов FMR1 заболевание проявляется или нет в зависимости от активности измененной хромосомы. Мужчины с ломкой хромосомой X не могут передать ее сыновьям, но передают всем дочерям, которые либо болеют, либо остаются здоровыми носителями мутации. Женщины с дефектной хромосомой передают ее детям обоих полов с вероятностью 50%. Наследование синдрома учащается от поколения к поколению, этот феномен называется парадоксом Шермана.
Патогенез
При секвенировании FMR1-гена было выявлено, что основой симптоматики и цитогенетически определяемой ломкости хромосомы X является многократное увеличение количества единичных тринуклеотидов ЦГГ. Это приводит к подавлению транскрипции и последующему недостаточному производству белка FMR1, ответственного за развитие центральной нервной системы, а именно – за формирование аксонов и синапсов, появление и усложнение нейронных связей, успешность процессов обучения и запоминания.
Участок хромосом, подверженный структурным изменениям при наследственном синдроме Мартина-Белл, может находиться в четырех состояниях, характеризующихся различным удлинением повторяющихся последовательностей тринуклеотидов. При отсутствии болезни и носительства определяется нормальное количество повторов – от 6 до 39. В промежуточном состоянии диагностируется 40-60 повторов, в состоянии премутации – 55-200. В обоих случаях заболевание отсутствует. Поскольку экспансия тринуклеотидов возможна лишь в период гаметогенеза, премутация способна превратиться в полную мутацию. Это происходит при передаче измененного материнского гена, аллель «утяжеляется» во время овогенеза. При полной мутации выявляется больше 200 повторов ЦГГ, чаще всего – от 230 до 4 000.
Симптомы
Дети рождаются с увеличенной массой тела, в среднем – 3,5-4 кг. Первыми обращают на себя внимание фенотипические особенности младенцев. Характерен макроорхизм – увеличение яичек без эндокринного заболевания. Окружность головы больше нормы или соответствует ее верхним границам. Лоб высокий и широкий, лицо вытянутое с уплощенной средней частью. Нос имеет слегка клювовидный загиб, ушные раковины крупные, располагаются низко. Суставы отличаются хорошей подвижностью, кости кистей и стоп широкие. Кожа зачастую гиперэластичная, волосы и радужные оболочки глаз светлого оттенка. Фенотипические признаки могут быть выражены по-разному, от одного-двух едва определяемых до полного комплекса.
Ключевое клиническое проявление заболевания – умственная отсталость. Стойкое интеллектуальное снижение проявляется слабым развитием сложных форм мышления и памяти. Пациентам недоступно понимание абстрактно-логических высказываний и явлений, использование категорий, установление аналогий. Сравнение, анализ и обобщение могут осуществляться на простом уровне, например, в конкретных бытовых ситуациях. Словарный запас обеднен. У многих мальчиков IQ равен 40-50 баллам, реже достигает 70-79. Относительно сохранна номинативная речь и зрительное восприятие. У девочек когнитивное снижение менее выраженное, соответствует легкой степени олигофрении или пограничному уровню интеллектуального развития.
Другой типичный симптом заболевания – своеобразие речи. Она ускоренная, сбивчивая, изобилует повторами, эхолалиями и персеверациями. Аутистические расстройства представлены трудностями коммуникации и поведенческими нарушениями. Дети часто проявляют агрессивность и замкнутость при попытке установления контакта. В тяжелых случаях развивается мутизм – полное отсутствие речи как средства общения. В поведении преобладает двигательная расторможенность, гиперактивность, стереотипии, самопровреждения. Пациенты избегают смотреть в глаза, не допускают прикосновений, но по сравнению с больными аутизмом интерес к общению присутствует. Стереотипные движения включают хлопки руками, прыжки, вращения вокруг своей оси, встряхивания руками, бег по кругу, гримасничанье и однообразное хныканье. Имеются трудности планирования и контроля поведения, переключения внимания и пространственной координации.
Неврологические симптомы неспецифичны. Определяется легкое снижение мышечного тонуса, двигательная дискоординация. Недостаточное развитие мелкой моторики затрудняет освоение письма, некоторых игровых и бытовых навыков (сборки конструктора, рисования, шитья и др.). У части больных имеются глазодвигательные нарушения, усиление сухожильных рефлексов, экстрапирамидные паракинезы, например, зажмуривание глаз, нахмуривание бровей, гримасничанье. При тяжелых формах синдрома возникают эпилептические припадки. У 25% пациенток с премутационным состоянием развивается первичная недостаточность яичников.
Диагностика
При выраженных фенотипических изменениях заболевание может быть обнаружено с первых месяцев жизни ребенка – неонатологи и врачи-педиатры обращают внимание на увеличенные размеры яичек и характерные особенности лица. В иных случаях подозрение на умственную отсталость возникает в возрасте от полугода до 2-3 лет. В этот период прослеживается отставание умственного развития, поведенческие и речевые нарушения. Дифференциальная диагностика нацелена на исключение РАС, в частности раннего детского аутизма, а также умственной отсталости другого происхождения (не связанной с ломкостью хромосомы Х). Обследование проводится психиатрами, неврологами и врачами-генетиками, включает:
- Клинический опрос, осмотр. В беседе с ребенком на первый план выходит снижение интеллекта, гиперактивность и расторможенность поведения, нарушение коммуникативных навыков. Уровень психического развития не соответствует возрасту, методики исследования интеллекта выявляют олигофрению (IQ – 40-79 баллов). Внешне наблюдаются характерные фенотипические признаки, при неврологическом осмотре выявляется мышечный гипотонус, усиленные сухожильные рефлексы, паракинезы.
- Генеалогический анализ. В отличие от других форм олигофрении при синдроме Мартина-Белл прослеживается наследственная передача болезни. Как правило, у пациента имеются родственники с данным заболеванием, чаще – мужчины (дед, дядя, брат). Иногда признаки легкого интеллектуального снижения обнаруживаются у матери, но диагноз у нее часто не установлен (не подтвержден).
- Генетическое исследование. В лабораторных условиях исследуется строение ДНК: определяется количество ЦГГ-повторов и статус метилирования. Применяется ПЦР и цитогенетический метод. Диагноз подтверждается, если количество триплетных повторов составляет более 200. При результате 60-199 возможны легкие фенотипические проявления болезни, риск развития патологии в следующем поколении (если показатель диагностирован у женщины).
Лечение синдрома Мартина-Белл
Методы специфической терапии синдрома в настоящее время отсутствуют. Проводится симптоматическое медикаментозное лечение и психолого-педагогическая коррекция. Усилия врачей и специальных психологов направлены на минимизацию эмоционально-поведенческих отклонений, овладение навыками ходьбы, речи и общения, чтения и письма. Медикаментозная терапия включает прием психостимуляторов, антидепрессантов, ноотропов, противоэпилептических средств и гормональных препаратов (при первичной недостаточности яичников). Обучение пациентов проводится по специальным коррекционно-развивающим программам. Для улучшения социальных навыков используются методы когнитивно-поведенческой терапии, групповые тренинги.
Прогноз и профилактика
Синдром Мартина-Белла не имеет осложнений и не сокращает продолжительность жизни больных, поэтому при своевременной и адекватной медико-психолого-педагогической помощи прогноз достаточно благоприятный: пациенты осваивают навыки общения и самообслуживания, обучаются в специальных школах, иногда овладевают рабочими профессиями. Профилактика основана на медико-генетическом консультировании пар из групп риска и пренатальной диагностике синдрома. Эти меры необходимы женщинам с синдромом преждевременного истощения яичников, семьям, в которых диагностированы премутационные состояния FMR1 или выявлены случаи интеллектуальной недостаточности у мальчиков и мужчин.
Источник
Синдром фрагильной Х-хромосомы (ломкой Х-хромосомы, ФХС, FRAX-А, синдром Мартина-Белла) – клиника, диагностикаСиндром фрагильной Х-хромосомы (синдром ломкой Х-хромосомы) (ФХС, иногда упоминается в литературе как «FRAX-А» и синдром Мартина-Белла) представляет собой сочетание соматических и поведенческих характеристик, связанных с фрагильным участком (и локусом гена) длинного плеча Х-хромосомы в области Xq27.3, который обнаруживается только в культуре с умеренным дефицитом фолата. Данное заболевание является второй после синдрома Дауна причиной задержки умственного развития с коэффициентом IQ менее 50 и наиболее частой причиной семейных случаев умственной отсталости и предпосылкой для развития различных поведенческих проблем, включая аутизм и синдром гиперактивности (Percy et al., 1990; Reiss и Freund, 1990; Hagerman и Hagerman, 2002). У многих пациентов отмечаются специфические нарушения речи, языковых навыков, поведения и социальных навыков. «Беспорядочная» речь является характерным признаком (Hanson et al., 1986). Типичны гиперактивность и дефицит внимания, а аутизм выявляется в 23% случаев (Bregman et al., 1987; Thake et al., 1987; Vieregge и Froster-Iskenius, 1989). Тем не менее, поведенческий фенотип отличается от проявлений аутизма, вызванного другими причинами, а гиперактивность может быть особенно ярко выражена (Baumgardner et al., 1995; Hagerman et al., 2005). Женщины, являющиеся носителями полной мутации одной из Х-хромосом, обычно имеют нормальный фенотип. Приблизительно в 35% случаев отмечается задержка умственного развития, обычно легкая, а у 15% пациенток отмечается пограничный уровень интеллекта, сложности в обучении или и то и другое (Kemper et al., 1986). Таким образом, ФХС является важной причиной легкой задержки умственного развития у женщин. Кроме того, приблизительно у 10% женщин с нормальным интеллектом, являющихся носительницами патологической хромосомы, могут отмечаться психиатрические заболевания, в особенности аффективное или шизоидное расстройство (Reiss и Freund, 1990). а) Патогенез. Синдром вызван увеличением «нормального» количества дупликаций тринуклеотида ЦГГ на участке Xq27.3 гена FMR1, кодирующего белок FMRR Имеются сообщения о развитии премутации при наличии более 55 повторов (51-54 повтора—пограничное число), а полная мутация отмечается, когда количество повторов превышает 200. Передача синдрома от мужчин без симптомов заболевания предполагает наличие премутаций, связанных с повтором 55-200 копий. Премутация имеет тенденцию к стабильности в процессе сперматогенеза, но часто превращается в полную мутацию в процессе овогенеза; таким образом, дочери мужчин-носителей всегда являются носительницами только премутации, в то время как у детей женщин-носительниц часто развивается клинически выраженная форма заболевания. У мужчин с полной мутацией практически всегда отмечается задержка умственного развития, в то время как у женщин это осложнение встречается лишь в 30-50% случаев и по сравнению с мужчинами менее выражено (Staley et al., 1993). Выявление фрагильного участка возможно только в 30-50% случаев. Присутствие полной мутации препятствует трансляции гена в белок (в результате метилирования) (Knight et al., 1993). Отсутствие гена FMRP в редких случаях регистрируется у пациентов без увеличения количества тринуклеотидов, а с множественными делециями или точечными мутациями гена FMR1 (Hirst et al., 1995). Зарегистрированы редкие случаи, когда у мужчин с полной мутацией сохранялись нормальные когнитивные способности (Smeets et al., 1995). Ген FMRP экспрессирован во многих тканях, с особым изобилием в нейронах. Его содержание у лиц с премутациями остается нормальным (Devys et al., 1993). Функции гена FMRP до сих пор до конца не изучены. Полная мутация гена может стать причиной аутизма, неспособности к обучению, тревожных расстройств и задержки умственного развития. Имеются данные о том, что аутизм также встречается у молодых мужчин, являющихся носителями премутантных аллелей. В ходе одного из недавних исследований было продемонстрировано, что приблизительно у 30% пациентов с ФХС отмечается аутизм; среди пациентов с аутизмом (по сравнению с пациентами только с ФХС) выявляются более низкие когнитивные способности, более выраженные проблемы с речью и отклонения поведения. Другие отклонения, связанные с премутантными формами гена, включают «синдром тремора-атаксии» (ФХТАС) среди пожилых мужчин и в редких случаях у женщин и преждевременную недостаточность яичников (Hagerman et al., 2005). У пациентов с синдромом фрагильной Х-хромосомы (ФХС) отмечается нарушение регуляции путей, связанных с метаботрофным рецептором глутамата-5; считается, что данное нарушение метаболизма обусловливает характерный фенотип.
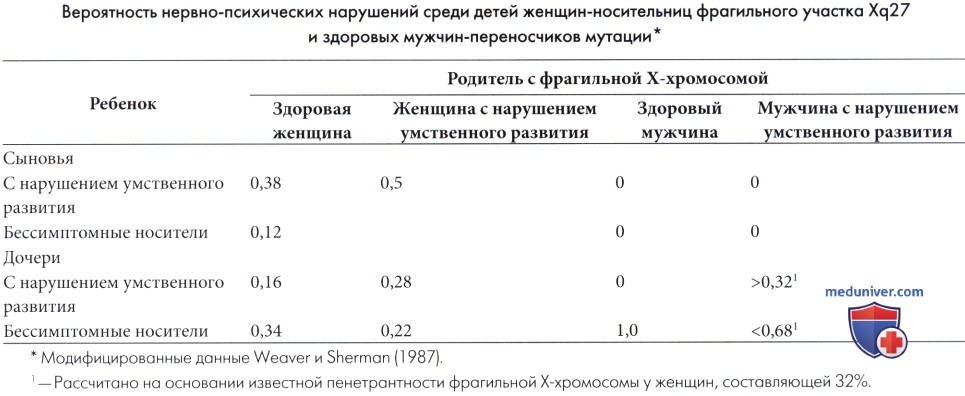
б) Распространенность. Синдром фрагильной Х-хромосомы (ФХС) (полная мутация ДНК) встречается с частотой приблизительно 1:4000 (среди мужчин) и 1:8000 (среди женщин), но учитывая, что исследования ДНК населения в целом не проводились, невозможно сделать вывод о достоверности данных сведений (Hagerman и Hagerman, 2002). Носительство премутаций отмечается с частотой 1:750-1000 мужчин и 1:250-350 женщин. С учетом того, что не у всех носителей фрагильной Х-хромосомы уровень IQ ниже 70, и они (не всегда) могут иметь лишь незначительные проблемы с учебой без гендерных различий, истинная распространенность ФХС (полной мутации и премутации с клинически значимыми отклонениями), вероятно, выше. У большинства мужчин с полной мутацией отмечаются отклонения от умеренной до тяжелой степени выраженности, в то время как у женщин проявления заболевания имеют менее выраженный характер, тем не менее, около трети пациенток имеют коэффициент IQ менее 70. Распространенность ФХС среди детей с трудностями в обучении от умеренной до тяжелой степени выраженности и без специфических дисморфических проявлений варьирует от 2% до 10% (Slaney et al., 1995). в) Диагностика. Диагноз синдрома фрагильной Х-хромосомы (ФХС) можно предположить на основании фенотипа; диагноз легко установить в постпубертатном периоде у мальчиков и затруднительно в препубертатном периоде у мальчиков и у девочек-носительниц, без типичных соматических изменений. Крупные уши и макроорхизм, напротив, часто не связаны с фрагильной Х-хромосомой (Hagerman, 1987). Семейный анамнез задержки умственного развития показателен, но обнаруживается только в трети случаев (Simko et al., 1989). Исследования ДНК в настоящее время надежны и оправдывают затраты (Rousseau et al., 1991, 1994; Oostra et al., 1993; Wang et al., 1993; Hagerman и Hagerman, 2002a). Экспресс-метод реакции антител, позволяющий выявить ген FMRP в лимфоцитах, был предложен в качестве скрининга для выявления полной мутации (Willemsen et al., 1995). Пренатальная диагностика синдрома фрагильной Х-хромосомы (ФХС) возможна с использованием культуры амниотических клеток или клеток ворсин хориона и методом анализа ДНК. Тем не менее, возникают этические проблемы, так как только у некоторых носителей мутации (особенно среди женщин) в дальнейшем разовьется заболевание. В настоящее время рассчитана (Weaver и Sherman, 1987) вероятность задержки умственного развития у детей больных родителей, но точные рекомендации остаются затруднительными.
г) Клинические проявления. Заболевание легче выявить у подростков, чем у детей в препубертатном возрасте. Наиболее важными проявлениями являются отсутствие замедленного физического развития, типично связанного с многими причинами умственной отсталости и, в особенности, нормальная или увеличенная окружность головы, вытянутое лицо с выступающей челюстью и макроорхизм (Но et al„ 1989). Классические проявления в виде вытянутого узкого лица и выступающих ушей часто незаметны в препубертатном периоде, хотя иногда бывают ярко выражены у молодых мужчин с ФХС. У взрослых женщин с полной мутацией часто отмечаются такие же изменения лица, но многие из них выглядят более «нормально», по сравнению с мужчинами. Макроорхизм отмечается приблизительно у трети мальчиков с ФХС и более чем у 90% взрослых мужчин. Другие частые изменения включают повышенную подвижность суставов пальцев, два сустава на первом пальце руки, плоские стопы, мягкую кожу, мышечную гипотонию, шум или дополнительный тон сердца (часто связанный с пролапсом митрального клапана или расширением корня аорты), косоглазие и высокое небо различной степени выраженности. В детском возрасте чрезвычайно часто встречается рецидивирующий средний отит, особенно по сравнению со здоровыми сибсами и детьми в общей популяции. В препубертатном возрасте у детей выявляются только следующие признаки: большая голова, большие уши и высокое небо. Помочь в установлении диагноза может также обнаружение повышенной гибкости суставов и мягкие уши. Несмотря на отсутствие макроорхизма у пациентов раннего возраста, в 15-50% случаев отмечается некоторое увеличение яичек (Simko et al., 1989). Типичные соматические проявления синдрома фрагильной Х-хромосомы (ФХС) представлены в таблице ниже. Задержка умственного развития при синдроме фрагильной Х-хромосомы (ФХС) обычно выражена слабо или умеренно, но с тенденцией к утяжелению в подростковом и взрослом возрасте (Borghgraef et al., 1987; Hagerman, 1987,1989; Wisniewski et al., 1989). У большинства мужчин по результатам стандартных тестов уровень IQ составляет менее 70 (чаще всего 35-40), но часть пациентов обладает нормальным или низко нормальным коэффициентом IQ. Вербальные навыки обычно превосходят способность к выполнению заданий и зрительно-пространственные навыки. Эпилепсия является достаточно частым проявлением и встречается в 15-25% случаев (Hagerman и Hagerman, 2002). Приступы часто имеют сложный парциальный характер, относительно доброкачественны и склонны к разрешению в подростковом возрасте, тем не менее, изредка отмечаются тяжелые и сложные случаи эпилепсии. Существует гипотеза о возможной связи припадков с аномалиями червя мозжечка, выявляемыми при ФХС. В подавляющем большинстве случаев у мужчин отмечаются явные признаки социальной дисфункции. Практически у всех мужчин присутствуют проявления аутизма, но лишь у немногих развивается полный синдром аутизма в сочетании с задержкой умственного развития или без нее. Фрагильная Х-хромосома наиболее распространена среди известных причин аутизма. Часто (приблизительно у половины мальчиков с полной мутацией) встречается гиперкинетический синдром в сочетании с аутизмом или без него, обычно с аутистическими проявлениями (Sullivan et al., 2006). Показатели когнитивных функций при ФХС отличаются от встречающихся при низкофункциональном аутизме, но достаточно часто соответствуют таковым при высокофункциональном аутизме и синдроме Аспергера. Некоторые авторы описывают сочетание синдрома Аспергера и синдрома фрагильной хромосомы (Hagerman, 1989). Некоторые варианты поведения, наиболее часто встречающиеся у пациентов мужского пола, включают избегание зрительного контакта, оборонительное поведение при прикосновении и социальную самоизоляцию (в возрасте от 0 до 2 лет); избегание зрительного контакта и приветственного поведения (отворачивание головы и тела при приветствии других людей), застенчивость, двигательные стереотипии различных видов и гиперактивность (в возрасте 3-4 лет); эхолалия, беспорядочная речь, «нервная непоседливость», хлопанье в ладоши, стереотипное махание вещами, кусание запястья или суставов пальцев, избегание зрительного контакта и приветственного поведения вместо стремления к социальной близости и заинтересованности в других людях (в возрасте 5-8 лет); сохраняющаяся застенчивость, избегание зрительного контакта и «нервозность», часто также привязанность к определенным вещам или людям на фоне общей картины умеренной задержки умственного развития с быстрой, беспорядочной речью с эхолалиями (очень часто отмечается разговор полушепотом, «нервный» смех) (в возрасте 9-12 лет); сохраняющиеся отклонения, часто усиливающиеся различными проблемами, связанными с началом пубертатного периода, включая трансвестизм, гипервозбудимость, самодеструктивное поведение и проблемы с одеждой (в связи с большим размером гениталий) (в возрасте 13-20 лет). В пубертатном периоде часто отмечается остановка когнитивного развития или даже ухудшение когнитивных функций. Сходная клиническая картина изредка отмечается у больных женщин, но в основном отклонения менее выраженные. У небольшого количества пациентов выявляется резко выраженный аутизм, а застенчивость и избегание зрительного контакта являются достаточно частыми проявлениями даже среди относительного большого числа больных без существенных отклонений. От трети до половины женщин с ФХС имеют различные варианты проблем обучения, которые варьируют от дислексии до легкой/умеренной задержки умственного развития. Имеются единичные сообщения о развитии шизоаффективного психоза у некоторых молодых женщин с фрагильной Х-хромосомой (Hagerman, 1989). У некоторых из этих женщин до развития психоза отмечались относительно небольшие проблемы в обучении, но с другой стороны, у них, по крайней мере, внешне, не выявлялось каких-либо проблем. Фенотип фрагильной Х-хромосомы также сочетается с синдромом тремора и атаксии (ФХТАС), который представлен тремором, атаксией, периферической поли-нейропатией и когнитивным дефицитом. Обычно отмечается значимая атрофия головного мозга и поражение белого вещества. ФХТАС развивается после подросткового возраста у мужчин (в отдельных случаях у женщин), имеющих премутации. Считается, что данный синдром связан с увеличением уровня аномальной информационной РНК гена FMR1. Имеющаяся в настоящее время информация в сочетании с недавно полученными сведениями о связи выраженности нейропатии (количества внутриядерных включений) и размера премутационного аллеля, подтверждает, что нейродегенеративный фенотип при ФХТАС является следствием распространенности повторов тринуклеотида ЦГГ (Cohen et al., 2006). Получены убедительные доказательства взаимосвязи премутации гена FMR1 и нарушения функции яичников с потерей фертильности (Wittenberger et al., 2007). Токсическое действие информационной РНК гена FMR1, кодирующей патологический белок, может являться причиной повреждения функции яичников. У женщин с преждевременной недостаточностью яичников отмечается повышенный риск наличия премутации гена FMR1; их информируют о возможности обследования на наличие фрагильной X-хромосомы. Специалисты по репродуктивной медицине могут обеспечить адекватные условия для разъяснения роли обследования на премутацию гена FMR1, создать условия для проведения и своевременно направлять пациенток на генетическую консультацию.
д) Лечение. В настоящее время специфического лечения синдрома фрагильной Х-хромосомы не существует (Hagerman, 1989). Некоторыми врачами использовалась фолиевая кислота (0,5-1,5 мг/кг, дважды в сутки), но эффективность сомнительна. В некоторых отчетах предполагается положительное воздействие фолиевой кислоты на симптомы аутизма, как минимум при назначении в дошкольном возрасте, но после пубертатного периода данный препарат оказывает незначительное отрицательное воздействие или не действует. Многие авторы использовали стимуляторы для подавления гиперактивности (в дозах, рекомендованных детям с синдромом дефицита внимания с гиперактивностью независимо от этиологии) и отмечали благоприятные или хорошие результаты (Hagerman и Hagerman, 2002). Ингибиторы обратного захвата серотонина могут быть эффективны в отношении тревожности, депрессивного настроения и раздражительности, но при применении необходим строгий контроль из-за возможности усиления импульсивности и агрессивного поведения. е) Другие, связанные с Х-хромосомой синдромы, с задержкой умственного развития. ФХС объясняется только 50% случаев преобладания задержки умственного развития среди мальчиков. Описано множество других более или менее идентифицированных синдромов связанной с Х-хромосомой задержки умственного развития (Opitz et al., 1986). Некоторые пациенты с анэуплоидией половых хромосом имеют трудности в обучении и другие нервно-психические отклонения. Данные заболевания описаны ниже. 1. FRAX-E. В некоторых случаях выявляется второй тип синдрома фрагильной Х-хромосомы (FRAX-E), вызванный распространением повторов тринуклеотида ГЦЦ в области Xq28, 600 тысяч нуклеотидов после гена FMR1 (Flynn et al., 1993). Задержка умственного развития обычно имеет слабо выраженный характер, а во многих случаях полностью отсутствует. 2. FRAX-F. Зарегистрирован третий вариант фрагильной Х-хромосомы (FRAX-F), который также связан с распространением повторов тринуклеотидов и может сочетаться с задержкой умственного развития и припадками (Hirst et al., 1993). 3. Синдром Ренпеннинга. Синдром Ренпеннинга, изначально считавшийся одним из клинических вариантов ФХС, характеризуется задержкой умственного развития от умеренной до тяжелой степени выраженности, легкой микроцефалией, низкорослостью и нормальным строением хромосом (Archidiacono et al, 1987). 4. Синдром Юберга-Марсиди. Данный редкий синдром включает такие проявления как задержка роста, глухота и микрогенитализм (Juberg и Marsidi, 1980). ж) Другие варианты рецессивных сцепленных с Х-хромосомой синдромов с задержкой умственного развития. Рецессивные сцепленные с Х-хромосомой синдромы с задержкой умственного развития были зарегистрированы также в сочетании с избыточным ростом тела (Golabi и Rusen, 1984), грубыми чертами лица, низкорослостью, макроорхизмом (Atkin et al., 1985) и прогрессирующими сложными неврологическими нарушениями (Schimke et al., 1984; Pfeiffer и Steffann, 1985). У родственников (мужского пола) пациентов со сцепленным с Х-хромосомой стенозом водопровода может отмечаться задержка умственного развития от умеренной до тяжелой степени выраженности, не сочетающаяся с гидроцефалией (Willems et al., 1987), иногда сопровождающаяся параплегией, приведенными первыми пальцами кистей и нарушениями речи (синдром задержки умственного развития-афазии-шаркающей походки-приведенных первых пальцев кистей). Сцепленная с Х-хромосомой глухота вследствие поражения слухового нерва, атрофия зрительного нерва и деменция могут быть проявлениями специфического рецессивного сцепленного с Х-хромосомой синдрома (Jensen 1981). – Также рекомендуем “Синдром Гольденхара – клиника, диагностика” Редактор: Искандер Милевски. Дата публикации: 4.12.2018 Оглавление темы “Наследственные синдромы в неврологии.”:
|

Источник