
Хромосомные болезни классификация и синдромы

3.1. Классификация хромосомных болезней
Хромосомные болезни (синдромы) – это группа врожденных патологических состояний, проявляющихся аномалиями развития и обусловленных нарушениями числа или структуры соматических хромосом (аутосомные синдромы) или половых хромосом (гоносомные синдромы). Их общая частота в популяции – около 1 %. В своем большинстве это спорадические случаи вследствие разнообразных хромосомных и геномных мутаций. Имеет право на существование и гипотеза о генетической детерминированности хромосмных нарушений. Число описанных типов хромосомных аномалий приближается к 1000, из них более 100 форм имеют клинически очерченную картину и называются синдромами [2].
Классификация хромосомных заболеваний основана на нескольких принципах:
I. Этиологический, т.е. характеристика хромосомной или геномной мутации:
1) Хромосомные болезни, связанные с аномалиями числа хромосом при сохранении их структуры.
– Болезни, обусловленные числовыми аномалиями половых (Х и Y) хромосом (болезни Шерешевского-Тернера, Клайнфельтера).
– Болезни, обусловленные числовыми аномалиями аутосом (синдромы Дауна, Патау, Эдвардса).
– Болезни, обусловленные увеличением кратности полного гаплоидного набора хромосом – полиплодии.
2) Хромосомные болезни, обусловленные структурными перестройками хромосом.
II. Определение типа клеток, в которых возникла мутация (в гаметах или зиготе):
– Гаметические мутации ведут к полным формам хромосомных болезней. У таких индивидов все клетки несут унаследованную с гаметой хромосомную аномалию.
– Соматические мутации – если аномалия возникает в зиготе или на ранних стадиях дробления, при этом развивается организм с клетками разной хромосомной конституции (два типа и более). Это явление называется мозаицизм, а формы хромосомных болезней – мозаичными. Для того, чтобы мозаичная форма по клинической картине совпадала с полной, необходимо иметь не менее 10 % клеток с аномальным набором.
III. Время возникновения мутации (в поколении):
– Спорадические случаи – мутация возникла заново в гаметах здоровых родителей или на стадии зиготы.
– Наследуемые (семейные) формы – когда родители уже имели подобную аномалию.
Таким образом, для точной диагностики хромосомной болезни необходимо определить:
1) тип мутации;
2) вовлеченную в процесс хромосому;
3) форму (полная или мозаичная);
4) вид болезни (спорадический случай или наследуемая форма).
Такая диагностика возможна только при цитогенетическом исследовании, проводимом у пациента, а иногда и у его родителей и сибсов. Диагностические признаки хромосомных синдромов можно разделить на три группы:
1. Общие признаки, позволяющие заподозрить аномалии хромосом (психическое или физическое недоразвитие, черепно-лицевой дисморфизм, пороки внутренних органов).
2. Признаки, чаще всего встречающиеся при определенных синдромах. Например, при синдроме Эдвардса в 90 % случаев встречается долихоцефалия и в 96 % – флексорное сгибание кисти. При синдроме Патау в среднем в 70 % случаев встречаются расщелина губы и нёба, микрофтальмия, поликистоз почек, полидактилия. При синдроме Дауна в более 90 % случаев отмечается монголоидный разрез глаз и в 60 % – поперечная складка на ладони.
3. Признаки, патогномоничные для определенного синдрома. Например, при синдроме Лежена отмечается характерный крик, напоминающий кошачье мяуканье, при синдроме де Груши – характерная алопеция.
Факторы повышенного риска рождения детей с хромосомными болезнями:
1. Потомство с трисомией появляется у одних и тех же женщин повторно с частотой не менее 1 %.
2. Родственники пробанда с трисомией 21 или другими анеуплоидиями имеют несколько повышенный риск рождения ребенка с анеуплоидией.
3. Кровное родство родителей может повысить риск трисомии у потомства.
4. Резко повышается риск рождения ребенка с трисомией у матери, чей возраст превышает 35 лет. После 45 лет каждая 5 беременность завершается рождением ребенка с хромосомной болезнью.
Источник
Хромосомные
болезни, или синдромы – это группа
врожденных патологических состояний,
проявляющихся множественными пороками
развития, различающихся по своей
клинической картине, часто сопровождающихся
тяжелыми нарушениями психического и
соматического развития. Основной дефект
– различные степени интеллектуальной
недостаточности, что может осложняться
нарушениями зрения, слуха, опорно-двигательного
аппарата, более выраженными, чем
интеллектуальный дефект, расстройствами
речи, эмоциональной сферы и поведения.
Диагностические
признаки хромосомных синдромов можно
разделить на три
группы:
неспецифические,
т.е. такие, как выраженная умственная
отсталость,
сочетающаяся с
дисплазиями, врожденными пороками
развития и черепно-лицевыми аномалиями;признаки,
характерные для отдельных синдромов;патогномоничные
для конкретного синдрома, например,
специфический плач при синдроме
«кошачьего крика».
Хромосомные
заболевания не подчиняются менделеевским
закономерностям передачи заболевания
потомству и в большинстве случаев
обнаруживаются спорадически, являясь
следствием мутации в половой клетке
одного из родителей.
Хромосомные
болезни могут быть унаследованы, если
мутация имеется во всех клетках
родительского организма.
К
механизмам, лежащим в основе геномных
мутаций, относятся:
нерасхождение
– хромосомы, которые должны были
разделяться во
время клеточного
деления, остаются соединенными и
относятся к одному полюсу;«анафазное
отставание» – утрата отдельной хромосомы
(моносомия)
может иметь место во время
анафазы, когда одна хромосома может
отстать от остальных;полиплоидизация
– в каждой клетке геном представлен
более чем
дважды.
Факторы, повышающие риск рождения детей с хромосомными болезнями
Причины
возникновения хромосомных болезней до
настоящего времени недостаточно изучены.
Имеются экспериментальные данные о
влиянии на мутационный процесс таких
факторов, как: действие ионизирующих
излучении, химических веществ, вирусов.
Другими причинами нерасхождения хромосом
могут быть: сезонность, возраст отца и
матери, порядок рождения детей, прием
лекарств во время беременности,
гормональные нарушения, алкоголизм и
др. Не исключается до определенной
степени и генетическое детерминирование
нерасхождения хромосом. Повторим,
однако, что причины образования геномных
и хромосомных мутаций на ранних стадиях
развития зародыша до сих пор окончательно
не раскрыты.
К
биологическим факторам повышения риска
рождения детей с хромосомными
аномалиями может быть отнесен возраст
матери. Риск рождения больного ребенка
особенно резко возрастает после 35 лет.
Это характерно для любых хромосомных
болезней, но наиболее четко наблюдается
для болезни Дауна.
В
медико-генетическом планировании
беременности особое значение уделяется
двум факторам — наличию анеуплоидии
по аутосомам у ребенка и возрасту матери
старше 35 лет.
К
кариотипическим факторам риска у
супружеских пар относятся: анеуплоидия
(чаще в мозаичной форме), робертсоновские
транслокации (слияние двух телоцентрических
хромосом в области деления) кольцевые
хромосомы, инверсии. Степень повышения
риска зависит от типа хромосомных
нарушений.

Синдром
Дауна (трисомия по 21 паре хромосом)
Причина:Нерасхождение
21 пары аутосом, транслокация 21 аутосомы
на аутосому группы D
или G.
У 94% кариотип — 47 хромосом. Частота
проявления синдрома увеличивается с
возрастом матери.
Клиника:
Признаки,
позволяющие диагностировать заболевание,
в типичных случаях выявляются на самых
ранних этапах жизни ребенка. Малый рост
ребенка, маленькая круглая голова со
скошенным затылком, своеобразное лицо
– бедная мимика, косой разрез глаз со
складкой у внутреннего угла, нос с
широкой плоской переносицей, маленькие
деформированные ушные раковины. Рот
обычно полуоткрыт, язык толстый,
неповоротливый, нижняя челюсть иногда
выступает вперед. На щеках часто
отмечается сухая экзема. Обнаруживается
укорочение конечностей, особенно в
дистальных отделах. Кисть плоская,
пальцы рук широкие, короткие. В физическом
развитии отстают, однако не резко, но
нервно-психическое развитие замедленно
(плохо развита речь). С возрастом
выявляется ряд новых черт заболевания.
Голос грубеет, отмечается близорукость,
косоглазие, конъюнктивиты, неправильный
рост зубов, кариес.Слабо развита иммунная
система, инфекционные заболевания
протекают крайне тяжело и в 15 раз чаще,
чем у других детей. Встречается острый
лейкоз.
Патогенез:Патологии
внутренних органов, сердечно-сосудистые
дефекты.
Диагностика:Клиническое
обследование, подтверждаемое
цитогенетическим анализом кариотипа.
Лечение:Комплексная
терапия, включающая правильную
организацию режима, рационально
построенная
медико-педагогическая работа, лечебная
физкультура,
массаж, медикаментозное лечение.

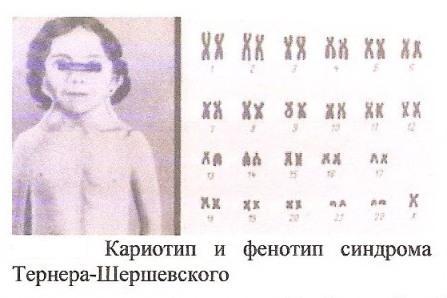
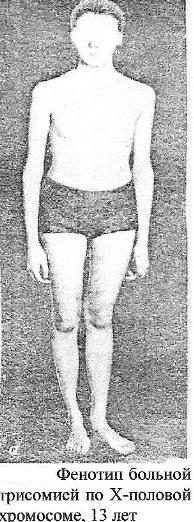
Синдром
Тернера-Шершевского (ХО)
Причина:Нерасхождение
половых хромосом, отсутствие одной
Х-хромосомы, кариотип – 45 хромосом.
Клиника:Низкий
рост,непропорциональное строение
тела, полная короткая шея с
крыловидными
кожными складками, широкая грудная
клетка, Х-образное искривление коленей.
Уши деморфированы, низко расположены.
Отмечается неправильный рост зубов.
Половой инфантилизм. Снижение умственного
развития.
Патогенез:
В пубертатный период недоразвитие
половых органов и вторичных половых
признаков, поражение сосудистой системы,
аномалии мочевой системы, уменьшение
остроты зрения, слуха.
Диагностика:
У
новорожденных ее установить трудно. С
возрастом диагностика основывается
на клинической картине и определении
патологии кариотипа и полового хроматина.
Лечение:
Симптоматическое, направленное на
увеличение роста. Для увеличения роста
используются анаболические гормоны. С
13-15 лет начинают лечение эстрогенными
препаратами. Полного выздоровления не
наблюдается, однако лечебные мероприятия
могут улучшить состояние
больных.

Синдром
Клайнфельтера (XXY;
XYY;
XYYYY;
XXXY)
Причина:Нерасхождение
половых хромосом, вследствие чего
увеличивается число X
или Y
хромосом в клетке, кариотип – 47 (XXY),
48 и более хромосом.
Клиника:Высокий
рост, отсутствие залысин на лбу, плохой
рост бороды, гинекомастия, остеохондроз,
бесплодие, слаборазвиты мышцы, аномалия
зубов и костной системы. Больные могут
демонстрировать сниженный интеллект.
С увеличением X-хромосом
увеличивается умственная отсталость
до полной идиотии, с увеличением
Y-хромосом
– агрессивность. Больные с более глубокой
степенью интеллектуального дефекта
могут обнаруживать ряд психопатологических
признаков: они мнительны, склонны к
алкоголизму, способны совершать различные
правонарушения.
Патогенез:В
пубертатном периоде обнаруживается
недоразвитие первичных половых признаков.
Диагностика:Основана
на клинических данных, а также на
определении патологического кариотипа
цитогенетическим методом, что
подтверждается исследованием полового
хроматина в клетках.
Лечение:Терапия
с помощью мужских половых гормонов для
увеличения потенции. Психотерапия.
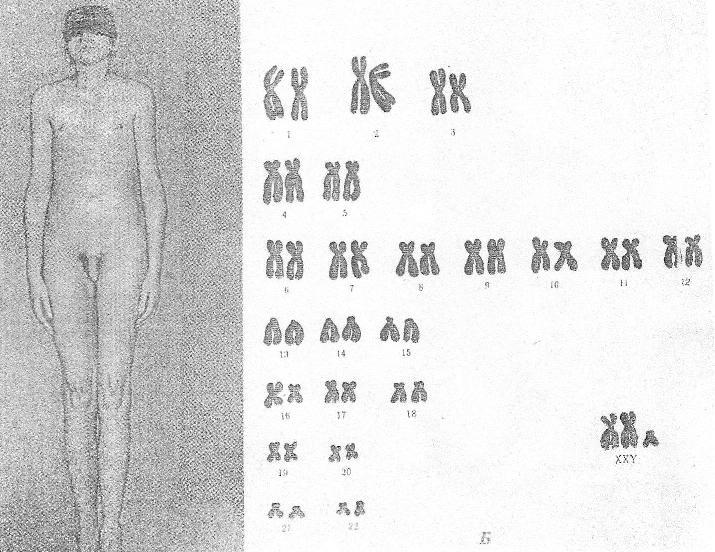
Синдром
Волъфа-Хиршхорна
Причина:
У
80 % страдающих им новорожденных
цитологическую основу данного синдрома
составляет деления короткого плеча 4-й
хромосомы. Размеры делеции колеблются
от небольших терминальных до занимающих
около половины дистальной части короткого
плеча. Отмечается, что большинство
делеции возникает заново, около 13 %
происходит, в результате транслокаций
у родителей. Реже в геноме больных,
помимо траснлокации, имеются и кольцевые
хромосомы. Наряду с делениями хромосом,
патология у новорожденных может быть
обусловлена инверсиями, дупликациями,
изохромосомами.
Клиника:
У
новорожденных небольшой вес при
нормальной продолжительности беременности.
Также отмечаются микроцефалия, клювовидный
нос, эпикант, антимонголоидный разрез
глаз (опущение наружных углов глазных
щелей), аномальные ушные раковины,
расщелина верхней губы и неба, маленький
рот, деформация стоп и др. Дети с синдромом
Вольфа-Хиршхорна маложизнеспособны,
как правило умирают в возрасте до одного
года.
Патогенез:
Болезнь характеризуется многочисленными
врожденными пороками развития, задержкой
умственного и психомоторного развития.
Диагностика:
По клинической картине.
Лечение:
Не
существует.
Синдром
трисомии (XXX)
Причина:Нерасхождение
половых хромосом в результате нарушения
работы митотического веретена деления
во время мейоза, кариотип — 47 хромосом.
Клиника:Пузырное
нерасхождение плаценты; новорожденный
имеет небольшой, широкий задний родничок,
недоразвитые затылочные и теменные
кости черепа. Отставание в развитии на
6-7 месяцев. Низко расположены деформированные
ушные раковины. Синдактилия пальцев
кисти, расщелина губы и неба, гидроцефалия.
Многие женщины нормально развиты,
интеллект ниже среднего. Частота развития
шизофреноподобных психозов увеличивается
второе.
Патогенез:
Пороки
развития внутренних органов.
Диагностика:По
клинической картине и цитогенетическому
определению патологии кариотипа и
полового хроматина.
Лечение:Симптоматическое.

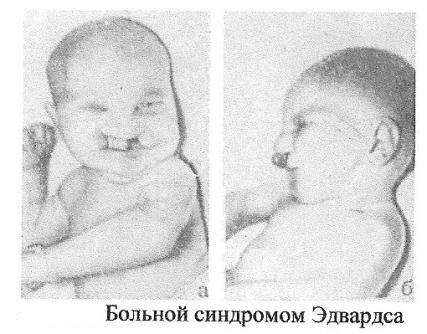
Синдром
Эдвардса (трисомия по 18 паре хромосом)
Причина:Нерасхождение
аутосом на стадии гамет (иногда зигот).
Лишняя
хромосома в 18 паре. Кариотип 47, Е18+.
Выражена зависимость
частоты рождения больных детей от
возраста родителей.
Клиника:
Пренатальное
недоразвитие, слабая активность плода,
нарушения
строения лица (короткие глазные щели,
маленькая верхняя челюсть)
и костно-мышечной системы практически
постоянны. Ушные раковины
деформированы и в подавляющем большинстве
случаев расположены
низко. Грудина короткая, ядра окостенения
расположены
неправильно и в меньшем количестве.
Спинномозговые грыжи и
расщелины губ.
Патогенез:Наиболее
постоянны пороки сердца и крупных
сосудов. Нарушения
развития головного мозга, в основном
гипоплазия мозжечка и мозолистого
тела. Из пороков глаз чаще всего
обнаруживается микроанафтольмия.
Врожденное отсутствие щитовидной железы
и надпочечников.
Диагностика:Клинический
осмотр,
дерматоглифика,
цитогенетическое
обследование.
Лечение:Отсутствует,
90% детей умирают на первом году жизни.
Выжившие
дети умирают от инфекционных заболеваний,
чаще от пневмонии.
Синдром
Патау (трисомия но 13 таре аутосом)
Причина:Нерасхождение
аутосом 13 пары в гаметогенезе у одного
из родителей. Кариотип – 47, D13+
.
Клиника:Аномалии
черепа и лица, окружность черепа
обычно уменьшена, в ряде случаев имеется
выраженная тригоноцефалия. Умеренная
микроцефалия сочетается со сравнительно
низким и скошенным лбом, узкими глазными
щелями, запавшим предносьем с широким
основанием носа, низко расположенными
и деформированными ушными раковинами.
Расстояние между глазными щелями часто
уменьшено. На коже головы имеются дефекты
скальпа овальной или круглой формы.
Часто – заячья губа и волчья пасть.
Аномалии костно-мышечной системы,
полидактилия.
Патогенез:Смертность
в течение первого года жизни (90%). Основной
причиной смерти детей являются тяжелые,
несовместимые с жизнью пороки развития:
дефекты сердечно-сосудистой и мочеполовой
систем, аномалии толстой кишки,
пупочная грыжа, нарушения строения
глазных яблок, постоянны микроанофтальмия,
дисплазия сетчатки, катаракты. Врожденные
пороки сердца встречаются у 80% детей.
Диагностика:Основана
на клиническом, цитогенетическим
исследованиях.

Синдром
“кошачьего крика”
Причина:Делеция
короткого плеча хромосомы 5-й пары.
Кариотип 46, 5р-.
Клиника:Патологическое
строение голосовых связок – сужение,
мягкость хрящей, отечность и
необычная складчатость слизистой,
мяуканье кошки. Недоразвитие речи.
Микроцефалия. Лунообразное лицо,
монголоидный разрез глаз, косоглазие,
катаракта, атрофия зрительного нерва,
плоская спинка носа, высокое нёбо,
деформированные ушные раковины.
Косолапость. Задержка умственного и
физического развития. Продолжительность
жизни значительно снижена, только около
14% больных переживают возраст 10 лет.
Патогенез:Порок
сердца.
Диагностика:Клиническое
обследование с выявлением наиболее
постоянного признака синдрома – “кошачий
крик”, дерматоглифика и цитогенетическое
выявление патологии кариотипа.
Лечение:Отсутствует.
Синдром
Орбели
Причина:
Деления
длинного плеча аутосомы 13.
Клиника:
Лоб
переходит в нос, не образуя носовой
вырезки. Большое расстояние между
глазами. Широкая спинка носа, высокое
нёбо, низко расположенные диспластичные
ушные раковины, пороки развития глаз
(косоглазие, катаракта). Пороки
опорно-двигательного аппарата
-неспецифические аномалии (косолапость,
вывих тазобедренных суставов). Задержка
роста и психомоторного развития;
характерна глубокая олигофрения. Больные
с развернутой клинической картиной
синдрома погибают на первом году жизни.
Патогенез:
Аномальное развитие практически всех
органов и систем; микроцефалия; врожденные
пороки сердца и аномалии прямой кишки.
Диагностика:
Цитогенетическое,
клиническое обследование.
Лечение:
Отсутствует.
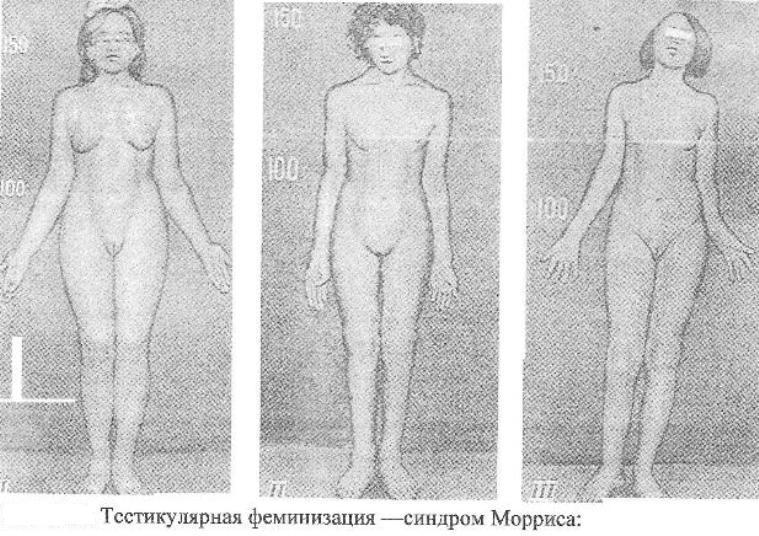
Синдром
Мориса
Причина:Мутация
гена, нарушающая образование нормального
белка — рецептора, делает ткани-мишени
невосприимчивому гормону, направляющему
их развитие по мужскому типу. Не
использовав такую возможность на
определенном этапе онтогенеза, организм
осуществляет развитие по женскому типу.
Клиника:Появляется
особь с кариотипом XY,
но внешне более сходна с женщиной. Такие
субъекты не способны иметь потомство,
так как их половые железы (семенники)
недоразвиты, а их выводные протоки часто
формируются по женскому типу (недоразвитая
матка, влагалище). Вторичные половые
признаки также характерны для женского
пола.
Патогенез:
Недоразвитые
половые органы.
Диагностика:
Цитогенетическое,
клиническое обследование.
Лечение:
Гормональная терапия.
Источник
Хромосомные болезни (греческий chroma цвет, окраска + soma тело) — группа болезней, в основе развития которых лежат нарушения числа или структуры хромосом, возникающие в гаметах (зрелых половых клетках) родителей или на ранних стадиях дробления зиготы (оплодотворенной яйцеклетки).
История изучения хромосомных болезней берет начало с клин, исследований, проводившихся задолго до описания хромосом человека (см. Хромосомы) и открытия хромосомных аномалий. Некоторые хромосомные болезни (например, болезнь Дауна) были описаны клиницистами как самостоятельные нозологические формы еще до раскрытия их хромосомной природы.
С 1959 года, когда франц. исследователи Ж. Лежен, Готье (М. Gauthier) и Тюрпен (R. Turpin) впервые описали нарушение кариотипа при болезни Дауна (см. Дауна болезнь, Кариотип), началось быстрое развитие клинической цитогенетики человека. Вскоре была раскрыта этиология синдромов Шерешевского — Тернера (см. Тернера синдром), Клайнфелтера (см. Клайнфелтера синдром), синдромов Патау (см. Патау синдром) и Эдвардса (см. Эдвардса синдром).
С разработкой методов авторадиографии (см.) стала возможной идентификация некоторых индивидуальных хромосом, что способствовало открытию группы хромосомных болезней, связанных со структурными перестройками хромосом — синдромы делеции (см.) коротких плеч 4-й и 5-й хромосом, синдром делеции 13-й хромосомы и др. В течение нескольких лет было открыто более десяти хромосомных болезней, детализированы особенности проявлений каждой из них на клиническом, цитологическом, биохимическом уровнях, определена их частота.
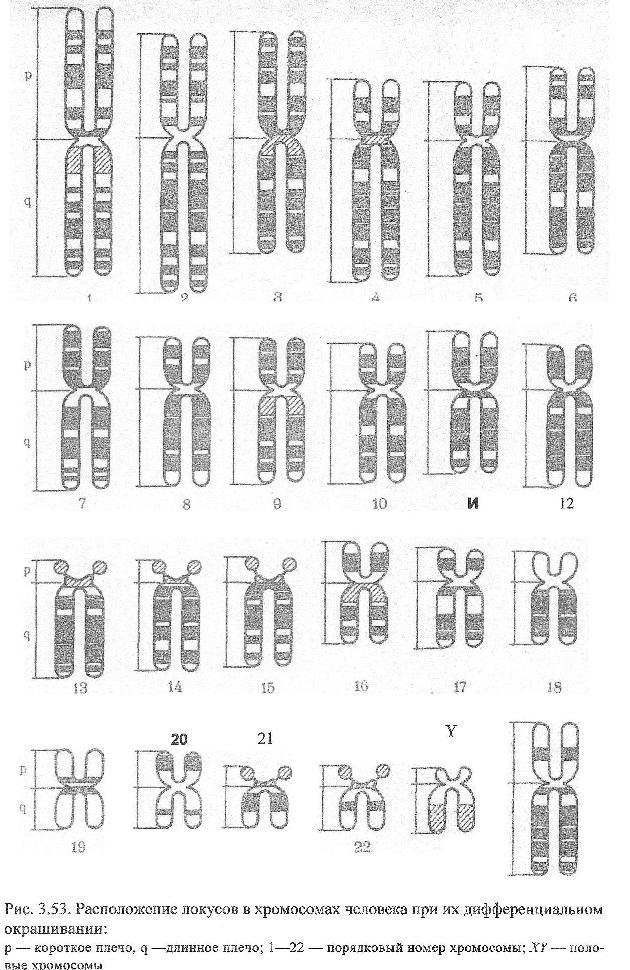
Интенсивное развитие учения о хромосомных болезнях началось в 70-х годах 20 века после разработки методов дифференциального окрашивания хромосом, что позволило точно идентифицировать индивидуальные хромосомы человека и даже их участки. Это обеспечило быстрое накопление клинико-цитогенетического материала и вычленение новых форм хромосомных болезней. В настоящее время известна клинико-цитогенетическая характеристика около ста хромосомных болезней и синдромов.
Классификация хромосомных болезней основана на типах мутаций (см. Мутация) вовлеченных в них хромосом. Мутации в половых клетках приводят к развитию полных форм Хромосомные болезни при которых все клетки организма имеют одну и ту же хромосомную аномалию. Мутации, возникшие в эмбриогенезе (особенно на начальных стадиях дробления зиготы), обусловливают возникновение мозаичных форм, при которых только часть клеток имеет хромосомную аномалию (см. Мозаицизм).
У человека обнаружены все основные формы и типы хромосомных аномалий. В настоящее время описано 2 варианта нарушений числа хромосомных наборов (см. Хромосомный набор) — тетраплоидия и триплоидия как в виде полных форм, так и мозаичных. Другая группа синдромов обусловлена нарушениями числа отдельных хромосом — трисомиями (когда имеется добавочная хромосома в диплоидном наборе) или моносомиями (одна из хромосом отсутствует). Моносомии аутосом (любые хромосомы, кроме X- и Y-хромосом) несовместимы с жизнью. Трисомии — более часто встречающаяся патология у человека. Так, наблюдаются хромосомные болезни, вызванные трисомией аутосом: по 13-й паре хромосом — синдром Патау, по 18-й паре — синдром Эдвардса, по 21-й паре — болезнь Дауна; значительно реже встречаются хромосомные болезни, вызванные трисомиями 8,9, 10,14,16,22-й пар хромосом. Ряд хромосомных болезней связан с нарушением числа половых хромосом. Так, моносомия X-хромосомы (генотип ХО) лежит в основе синдрома Шерешевского — Тернера, трисомия половых хромосом (генотип XXY) является причиной развития синдрома Клайнфелтера.
Самая многочисленная группа хромосомных болезней — это синдромы, обусловленные структурными перестройками хромосом. Описано более 700 типов этих нарушений. Однако не все они могут рассматриваться как самостоятельные клинические синдромы, так как при единичных наблюдениях нельзя связывать клин, картину непосредственно с хромосомной аномалией как этиологическим фактором. Тем не менее только лишь по аутосомам можно выделить около 80 синдромов, вызванных перестройками, сопровождающимися либо утратой (делецией) части хромосомного материала, либо его избытком. В связи с этим выделяют хромосомные синдромы так называемых частичных моносомий и синдромы частичных трисомий (уменьшение или увеличение числа отдельных хромосом не на целую хромосому, а на ее часть).
В связи с тем, что подавляющая часть хромосомных аномалий относится к категории летальных мутаций, для характеристики их количественных параметров используются два показателя — частота распространения и частота возникновения.
Фактический материал цитогенетического обследования новорожденных, проведенного в нескольких странах (СССР, Дания, Шотландия, Канада, США), дает достаточно полное и точное представление об общей частоте хромосомных болезней в популяциях человека, которая достигает 1%. При этом частота отдельных типов составляет: аномалии в системе половых хромосом среди мальчиков — 0,3% (из них XXY—0,15%, XYY — 0,11%, структурные перестройки X-и Y-хромосомы — 0,04%); среди девочек — 0,22% (ХО полная форма — 0,01 %, ХО мозаичная форма — 0,07%, XXX — 0,14%); трисомии аутосом — 0,14% ( из них трисомия 13—0,007%, трисомия 18— 0,013%, трисомия 21—0,12%); структурные перестройки аутосом — 0,24% (робертсоновские транслокации — 0,1%; реципрокные транслокации — 0,09%; инверсии — 0,02%; дупликации — 0,03%).
Выяснено, что около 170 из 1000 эмбрионов и плодов погибают до рождения (подвергаются так называемому внутриутробному отбору, что клинически выражается спонтанным абортом или мертворождением), из них около 40% — вследствие влияния хромосомных нарушений. Тем не менее значительная часть мутантов (носителей хромосомной аномалии) минует действие внутриутробного отбора. В среднем у 7 из 1000 живорожденных детей определяются различные хромосомные аномалии. Вследствие тяжести морфологических, физиологических и биохимических нарушений большинство больных с синдромом Пата у и Эдвардса погибает в раннем возрасте, с болезнью Дауна — до достижения пубертатного возраста. Больные с аномалиями половых хромосом из-за нарушений полового развития и нередко выраженных эндокринопатий, как правило, не оставляют потомства. Следовательно, все аномалии кариотипа (числа хромосом) являются вновь возникшими и их можно отнести к мутациям, элиминирующимся в первом поколении, то есть они существуют в популяции с той частотой, с к-рой возникают. Действию естественного отбора на разных этапах онтогенеза подвергаются и аномалии структуры хромосом. Показано, что в общем случае хромосомные мутации почти полностью исчезают из популяции через 15—17 поколений после своего возникновения.
Для всех форм хромосомных болезней общим признаком является множественность морфологических, физиологических и биохимических нарушений. Основные клин, проявления характеризуются нарушениями морфогенеза в виде множественных врожденных пороков развития. Их формирование начинается со стадии гистогенеза и продолжается в органогенезе, что, вероятно, и объясняет сходство клинической картины при разных формах хромосомных болезней. Общими проявлениями разных хромосомных болезней являются: задержка физического и психомоторного развития, умственная отсталость различной степени выраженности, черепно-лицевые дисплазии, костномышечные аномалии, пороки сердечно-сосудистой, мочеполовой, нервной и других систем, отклонения в гормональном, биохимическом и иммунологическом статусе.
Степень поражения органов и систем при хромосомной болезни зависит от многих факторов — типа хромосомной аномалии, индивидуальности по генному составу вовлеченной в аномалию хромосомы, размера недостающего или избыточного материала индивидуальной хромосомы, степени мозаичности организма (см. Мозаицизм) по аномальному клеточному клону (см.), генотипа организма, условий среды, в которых развивается организм.
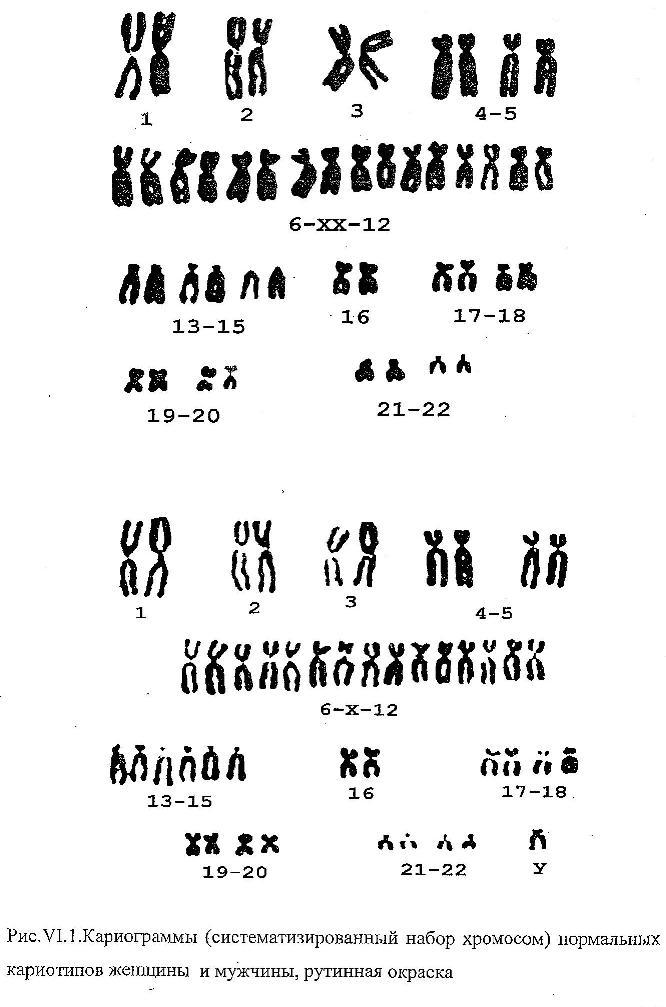
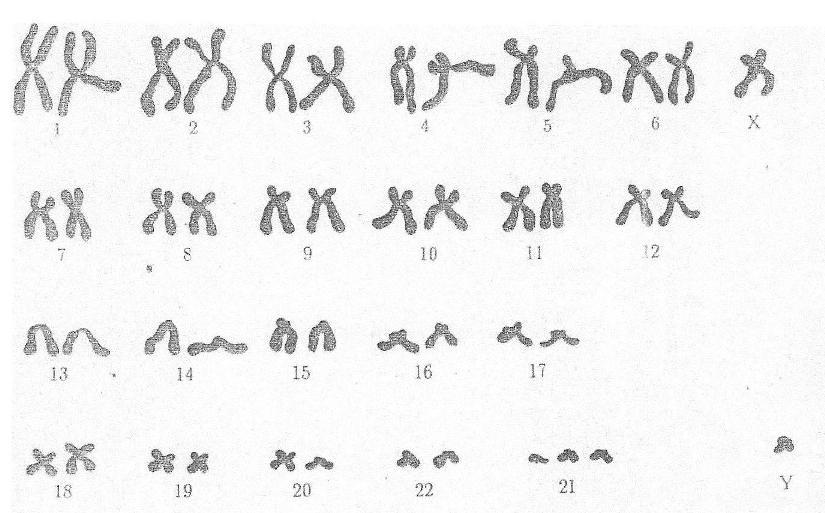
Диагностика хромосомных болезней основана на клинико-морфологических и цитогенетических методах. Нарушения морфогенеза, клинически определяемые как врожденные пороки развития, являются характерной чертой почти всех хромосомных болезней. При отдельных формах выявляется до 20 врожденных аномалий и пороков развития различных органов и систем. Одни признаки относительно постоянны для всех или большинства хромосомных аномалий, другие — встречаются только при определенных хромосомных болезнях, третьи — лишь при данной хромосомной болезни. Постановка диагноза по клин, картине не очень надежна и всегда требует цитогенетического подтверждения.
В зависимости от целей исследования применяют цитогенетические экспресс-методы — анализ X- и Y-хроматина (см. Половой хроматин) и методы точного хромосомного анализа со специальной дифференциальной окраской хромосом (см. Хромосомы). Выбор того или иного метода анализа хромосом зависит от цели исследования и требуемой степени точности. В случае необычной клинической картины синдромов с численными аномалиями хромосом или при подозрении на наличие структурной перестройки необходимо исследовать хромосомные наборы, используя несколько методов дифференциальной окраски хромосом. В ряде случаев для уточнения наличия микроперестройки или варианта хромосомы требуется применение высоко разрешающих методов анализа прометафазных или профазных хромосом.
Этиологическое лечение хромосомных болезней в настоящее время не разработано. Патогенетические методы лечения включают коррекцию метаболических и гормональных нарушений.
Симптоматическое лечение применяется практически при всех формах хромосомных болезней и включает физиотерапию, назначение витаминов и другие лечебные мероприятия. Необходимо при этом исключить факторы, усиливающие проявления хромосомных болезней, а также применять специальные методы воспитания и обучения, что позволяет во многих случаях обеспечить достаточную социальную адаптацию.
Прогноз при хромосомных болезнях зависит от тяжести поражения органов и систем, адекватности проводимых лечебных мероприятий. Основой профилактики хромосомных болезней является медико-генетическое консультирование, которое заключается в определении прогноза рождения ребенка с хромосомной патологией (см. Медико-генетическая консультация). Разработка методов пренатальной диагностики делает этот подход одним из самых эффективных в борьбе не только с хромосомными, но и с другими наследственными болезнями (см.).
Библиогр.: Бочков Н. П. Хромосомы человека и облучение, М., 1971; он же, Генетика человека, наследственность и патология, М.,1978; Бочков Н. П., 3ахаров А. Ф. и Иванов В. И. Медицинская генетика, М., 1984; Захаров А. Ф. и др. Хромосомы человека, Атлас, М., 1982; Перспективы медицинской генетики, под ред. Н. П. Бочкова, М., 1982; Тератология человека, под ред. Г. И. Лазюка, М., 1979; Hamerton J. L. Human cytogenetics, v. 1—2, N. Y.—L., 1971; Hamerton J. L. a. o. A cytogenetic survey of 14069 newborn infants, Clin. Genet., v. 8, p. 223, 1975; Jacobs P. A. a. o. A cytogenetic survey of 11680 newborn infants, Ann. hum. Genet., v. 37, p. 359, 1974; LejeuneJ., Gauthier M. et Turpin R. Les chromosomes humains en culture de tissus, C. R. Acad. Sci. (Paris), t. 248, p. 602, 1959; Nielsen J. a. Sillesen I. Incidence of chromosome aberrations among 11148 newborn children, Humangenetik, v. 30, p. 1, 1975.
H. П. Кулешов.
Источник