
Эпилепсия и синдром арнольда киари

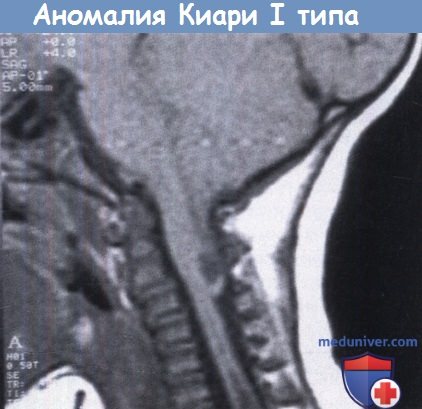
Алгоритм диагностики и лечения аномалии Киари 1 типа – Европейские рекомендацииВ последнем десятилетии XIX века немецкий патологоанатом Киари описал четыре врожденные аномалии мозжечка и ствола мозга, как проявления нарастающей тяжести единого патогенетического механизма, связанного с давлением сзади на задний мозг при врожденной гидроцефалии. Основной анатомической особенностью аномалии Киари I типа является разная степень каудального смещения миндалин мозжечка в шейный канал. Аномалия Киари 2 типа характеризуется более тяжелой мозжечковой грыжей, которая, кроме миндалин мозжечка, также включает в себя нижний червь и четвертый желудочек; она, как правило, связана с миеломенингоцеле. Аномалия Киари 3 типа включает в себя признаки высокого менингоэнцефалоцеле с грыжей заднего мозга. Наконец, Киари 4 типа характеризуется тяжелой гипоплазией мозжечка без каудального смещения. После многих лет и большого количества теоретических, клинических и нейровизуализационных исследований, понимание и осознание этого состояния вышло на новый уровень. В настоящее время считается, что эти четыре порока развития являются отдельными состояниями, с различным патогенезом, клиническим проявлением, и прогнозом, а не разными прогрессирующими стадиями одного заболевания. С анатомической точки зрения аномалии 1-3 типа являются грыжами заднего мозга различной степени, с возможным вторичным образованием полости в спинном мозге, в то время как аномалия 4 типа характеризуется гипоплазией мозжечка. Гипоплазия задней черепной ямки является частой находкой при первых трех аномалиях, но никогда не встречается при четвертой. У каждой формы свои патогномоничные клинические проявления и по этим причинам различные аномалии Киари должны рассматриваться в разных разделах. Мальформация Киари 1 типа. Аномалия Киари 1 типа характеризуется неправильным положением (и формой) миндалин мозжечка, которые опускаются из полости черепа ниже большого затылочного отверстия в шейный канал. Это анатомическое нарушение приводит к окклюзии субарахноидального пространства на уровне большого затылочного отверстия с последующим нарушением ликвороциркуляции в головном и спинном мозге; фактически, помимо изменений в задней черепной ямке, повышение ликворного давления на этом уровне приводит к сирингомиелии. Патогенетические механизмы развития аномалии детально изучены и обсуждены в литературе. Различные механизмы патогенеза можно схематично разделить на три категории: Хотя аномалия Киари 1 типа обычно становится симптоматичной в раннем подростковом периоде, в последнее время она обнаруживается с одинаковой частотой как у детей, так и у взрослых. а) Клиническая картина аномалии Киари I типа. Наиболее частой жалобой у этой группы пациентов является боль в затылке или в шее, усиливающаяся при чихании, кашле или при пробе Вальсальвы. Другими болезненными проявлениями являются боль в плече, спине или боли в конечностях без корешкового распределения. Наиболее частым симптомом являются признаки моторного или сенсорного дефицита в конечностях (> 70%), которые являются проявлением наличия полости в спинном мозге (сирингомиелии). Атаксия тела и конечностей в качестве проявление нарушений в мозжечке является вторым наиболее распространенным симптомом (в 30-40%). Реже (в 15-25%) наблюдается неуклюжесть, нистагм, диплопия, дисфагия и дизартрия как проявление дефицита черепных нервов. Апноэ при заикании наблюдается в 10% случаев, в большинстве случаев проявляясь у младенцев или маленьких детей. Своеобразным проявлением аномалии Киари 1 типа у детей и подростков является прогрессирующий сколиоз (в 30%). б) Лучевая диагностика. МРТ — лучший способ для диагностики аномалии Киари 1 типа. Важными критериями для этого порока являются: пролабирование одного или обоих миндалин мозжечка ниже большого затылочного отверстия (> 5 мм); возможны шейно-мозговая деформация, отсутствие супратенториальных нарушений (за исключением отдельных случаев небольшого расширения желудочков и обычная локализация четвертого желудочка. Аномалия Киари 1 типа может быть связана с такими костными аномалиями, как маленькая задняя черепная ямка, платибазия, атланто-затылочная ассимиляция, базилярная импрессия, срастание шейных позвонков (аномалия Клиппеля-Фейля). Гидро/сирингомиелия встречается в 50-60% случаев. Она может быть ограничена одной или двумя областями, а может распространяться на всю длину спинного мозга. Обычно полость образуется на уровне С1. МРТ является полезным дополнением к диагностическим мероприятиям, так как позволяет определить степень сдавления ствола на уровне затылочного отверстия и характеристики ликвородинамики; исследование, выполненное в послеоперационном периоде, позволяет получить некоторое представление об адекватности хирургической декомпрессии. в) Лечение аномалии Киари I типа. Целью хирургического лечения является декомпрессия задней черепной ямки для восстановления циркуляции спинномозговой жидкости в базальных цистернах и устранение сдавления нервных структур на уровне краниоцервикального перехода. Хирургическое лечение при аномалии Киари 1 типа обычно определяет четко определенный протокол, который включает субокципитальную краниотомию, С1 ламинэктомию, лизис арахноидальных сращений, резекцию миндалин мозжечка (как традиционную тонзилэктомию, так и субпиальную коагуляцию) и расширенную дуропластику. В случае сопутствующей вентральной компрессии (как, например, при платибазии, С1 ассимиляции и т.д.), она должна быть устранена до проведения дорсальной декомпрессии. В последнее время появляются сообщения, которые указывают на возможность достижения сопоставимых результатов при использовании простой декомпрессии, без расширяющейся дуропластики (или с отслоением наружного слоя твердой мозговой оболочки). Интраоперационная ультразвуковая диагностика может быть полезна в этом отношении, демонстрируя синхронизацию движения миндалин с дыханием и сердцебиением, а также наличие адекватного движения ликвора из четвертого желудочка. Дальнейшие хирургические возможности представлены процедурами, направленными на лечение гидро/сирингомиелии (закупорка задвижки, размещение сиринго-субарахноидального или сиринго-плеврального шунта, и стентирование четвертого желудочка). При сравнении двух основных крупных хирургических вмешательств при лечении аномалии Киари 1 типа с сирингомелией — субокципитальной декомпрессии и установки сиринго-субарахноидального шунта—Hida et al. обнаружили уменьшение размера сирингомиелитической полости у 94% пациентов, перенесших декомпрессию, и у 100% перенесших сирингосубарахноидальное шунтирование. Что касается прогноза, то максимальные преимущества декомпрессии проявляются у пациентов с симптомами, связанными с пароксизмальной внутричерепной гипертензией. Кроме того, пациенты с мозжечковой симптоматикой и синдромом большого затылочного отверстия имеют больше шансов на выздоровление, чем пациенты с центральным синдромом спинного мозга. При работе с пациентами детского возраста главной проблемой являются показания к операции. Фактически, во многих случаях диагноз аномалии Киари ставится случайно, после МРТ для диагностики причины неспецифических клинических проявлений, таких как головные боли, умственная отсталость, эпилепсия. Большинство авторов не согласно с превентивным хирургическим лечением, считая, что показания к нему должны основываться на наличии симптомов и клинических проявлений, однозначно относящихся к аномалии Киари, а не данными нейровизуализации. Что касается хирургической техники, некоторые авторы используют те же оперативные приемы, что и для взрослых, т. е. субокципитальную декомпрессию, С1 ламинэктомию, пластику ТМО и резекцию миндалин мозжечка в 40-60% случаев. При послеоперационной МРТ отмечается 100% улучшение симптомов, а также более чем 90% улучшение признаков, связанных с сирингомиелией и уменьшение сирингомиелитических кист в 80% случаев. В последние годы некоторые авторы критикуют этот «классический» подход как слишком тяжелый (а именно интрадуральные манипуляции), особенно в случаях умеренного опущения миндалин. В действительности, по некоторым сообщениям существуют указания на возможность достижения сопоставимых результатов менее инвазивным методом, то есть выполнением субокципитальной декомпрессии (и С1 ламинэктомия) с или без расслаивания внешнего слоя твердой мозговой оболочки. Улучшение или разрешение клинических проявлений описывается у более чем 90% детей, а исчезновение сирингомиелической полости — в 80% случаев. Удаление миндалин мозжечка с вскрытием ТМО или дуропластикой должно использоваться в случаях неэффективности минимально инвазивного лечения.
– Также рекомендуем “Алгоритм диагностики и лечения аномалии Киари 2 типа – Европейские рекомендации” Оглавление темы “Аномалия Киари.”:
|
Источник
Со степенями давайте не будем уточнять в названии, ок?
По просьбам.
Администрация.
Ветка про аномалию Арнольда-Киари (сдавливание мозжечка) https://vk.com/topic-248732_23599016
Операция в 12 лет https://vk.com/wall-248732_786529
Что такое синдром Арнольда-Киари?
В задней черепной ямке в норме располагаются такие важнейшие структуры, как часть полушарий, продолговатый мозг, миндалины мозжечка. В этой области присутствует переход головного мозга в спинной. Структуры спинного мозга переходят в область шейного позвоночного канала через затылочное отверстие.
синдром Арнольда-Киари
Мальформация Киари характеризуется появлением нарушений развития костей черепа.
https://mozgmozg.com/bolezni/sindrom-arnolda-kiari
Отредактировал администратор, 27 мая 2020 в 7:46.
мне после обследования поставили такой диагноз,рекомендована операция в питерском нии им.Поленова…боюсь…вся эта веселая вещь сопровождается “ложными” эпи-приступами с тонической судорогой…
может,у кого-то был сходный диагноз или кому-то уже проводили такую операцию?будьте добры,поделитесь,пожалуйста, опытом!расскажите,какие последствия принесет с собой операция и предстоит ли круто менять свою жизнь после этого?
Мужу в диагнозе ставят эпи и Аномалию Арнольда-Киари-подскажите кто нибудь что это за аномалия???????Заранее спасибо
а это может быть причиной эпиприступов?
у меня Аномалия, оперировали…через 4 года поставили эпилепсию. каких-то особых улучшений после операции я не наблюдала, разве что первые пол года.
если кому-то нужен совет из личного опыта, обращайтесь.
Мне тоже 2 года назад поставили этот диагноз по результатам МРТ! 3 степень, говорят не смертельно и жить можно…
Жить можно и нужно, я уже и забыл про неё, да вот на тему наткнулся )
Три года живу и ничего )
Тож на мрт появилась, диакарб ща пью, т. К желудочки могут сдавливать мозг в зоне ээг очага. Никак ее не ощущаю
Всем здравствуйте! у моего мужа киари 1 типа, ездили в Москву недавно, в Бурденко, до этого 3 года лечились и пытались найти выход, но увы врачи подтвердили ,что нужно делать операцию. Операция эта НЕСЛОЖНАЯ! студенты интернатуры проходят эту операцию как самую первейшую и лёгкую. Разрезается сзади черепа,чуть выше шеи, там расширяется так сказать отверстие для мозжечков , убирается одна косточка, вообщем есть видео на ютюбе, посмотрите если не страшно. Кто то живёт с этим всю жизнь, может появиться в 60 лет, в 35,а может вообще не появиться. У мужа была предрасположенность, после того как заклинело шею, отправился по молодости к мануалу, дак вот этот урод дёрнул ему шею без мрт , и мозжечки провалились! ДОРОГИЕ НИКОГДА НЕ ХОДИТЕ К МАНУАЛАМ!ОНИ ШАРЛАТАНЫ! И НЕ ОДИН ВРАЧ Н БУДЕТ ЧТО ТО ДЕЛАТЬ БЕЗ СНИМКА!ЕСЛИ БЕЗ НЕ ПРОСИТ СНИМОК ТО БЕГИТЕ ОТ ТУДА! вообщем 5 августа у нас госпитализация, будем делать операцию, муж жутко переживает, всё таки голова, если кто то делал то пожалуйста отзовитесь!
Виктория, у меня тоже киари.так у вашего мужа из-за этого приступы?может и у меня…
Появились в детстве приступы с судорогами, проходила разные обследования ЭЭГ, МРТ… В итоге поставили диагноз эпилепсия. А через 13 лет на очередном МРТ нашли как раз этот синдром. Одна “замечательная” врачиха сказала моим родителям что при таком состоянии, я могу просто гулять по улице и бамс мозжечок провалится в большую затылочную цистерну и я умру… Мне сразу через месяц сделали операцию. Правильно пишут: ничего страшного в ней нет, нужен только опытный нейрохирург. Надеялась что после операции приступы пройдут, но нет надежда не оправдалась… А насчёт операции если кого нужно успокоить – пишите л/с
Анна, напишите мне , про анамалию . У меня 1 степень , но операцию не предлагали
Гипокальциемия – потенциально угрожающее жизни состояние в виде снижения уровня кальция в сыворотке крови. Нормальные значения 2,1–2,6 ммоль/л, уровень ниже 2,1 ммоль/л определяется как гипокальциемия.
Клинические проявления гипокальциемии могут широко варьироваться – от асимптомных форм, выявленных при лабораторной диагностике, до угрожающих жизни метаболических нарушений. Гипокальциемия встречается при ряде заболеваний, которые можно разделить на нарушения секреции или действия паратгормона, нарушения,связанныесвитаминомД,атакже при патологии кальций-чувствительного рецептора. Более практичная классификация разделяет этиологию гипокальциемии поуровнюПТГвсыворотке–низкийилине определяющийся, нормальный, повышенный.
Гипокальциемия с низким или не определяющимся уровнем паратгормона (ПТГ) наблюдается при врожденном гипопаратиреозе, который может быть как изолированным, так и ассоциированным с другими дефектами развития. Наиболее известный синдром, ассоциированный с врожденным гипопаратиреозом, – синдром Ди Джорджи (синдром дизэмбриогенеза 3–4 жаберной дуги, врожденная аплазия тимуса и паращитовидных желез, синдром 22q11.2). Клинические проявления синдрома Ди Джорджи варьируются, наиболее постоянными являются гипопаратиреоз, кандидомикоз, гипокальциемическиесудорогивнеонатальномпериоде, иммунологическая недостаточность, черепно-лицевые дизморфии. Также низкий уровеньПТГнаблюдаетсяприаутоиммунном полигландулярном синдроме 1 типа в сочетании с хроническим кожно-слизистым кандидозом, надпочечниковой недостаточностью, алопецией.
При гипопаратиреозе могут встречаться разнообразные приступы (генерализованные тонические, сложные фокальные, абсансные, акинетические), являющиеся по своей природе гипокальцемическими. В литературе, посвященной гипопаратиреозу, имеются указания на «хорошую» адаптацию пациентов к гипокальцемии, когда судороги могут не возникать даже при низких показателях кальция в крови при длительном существовании данных изменений. При проведении МРТ головного мозга у пациентки была заподозрена фокальная корковая дисплазия, относящаяся к эпилептогенным дисгенезиям головного мозга, что заставляло думать о симптоматической фокальной эпилепсии. Настораживало, что иктальный очаг (правая лобно-височная область) не совпадал с «подозрительным» очаговым изменением коры головного мозга (левая височная область). Диагностика гипокальцемии носила, скорее, случайный характер и была выявлена при лабораторном контроле, который регулярно проводится у пациентов, получающих противосудорожную терапию.
https://lib.medvestnik.ru/articles/Epilepsiya-i-gipop..
Эпилепсия, это все МРТ регистрирует или здесь история генетическая?. Добрый день!
Есть ли смысл сдавать генетическую панель Эпилепсии (синдромы и т.д), все остальное уже проверено вроде…
Источник
Аномалия Киари (синдром Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
Общие сведения
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и др. аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Аномалия Киари
Причины возникновения аномалии Киари
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация аномалии Киари
Аномалия Киари подразделяется на 4 типа:
Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Симптомы аномалии Киари
Наиболее часто в клинической практике встречается аномалия Киари I типа. Она проявляется ликворногипертензионным, церебеллобульбарным и сирингомиелическим синдромами, а также поражением черепно-мозговых нервов. Обычно аномалия Киари I манифестирует в период полового созревания или уже во взрослом возрасте.
Для ликворногипертензионного синдрома, которым сопровождается аномалия Киари I, характерна головная боль в затылке и шейной области, усиливающаяся во время чихания, кашля, натуживания или напряжения мышц шеи. Может наблюдаться рвота, не зависящая от приема пищи и ее характера. При осмотре пациентов с аномалией Киари выявляется повышенный тонус мышц шеи. Среди мозжечковых нарушений наблюдаются нарушение речи (дизартрия), нистагм, мозжечковая атаксия.
Поражение ствола мозга, расположенных в нем ядер черепно-мозговых нервов и их корешков проявляются снижением остроты зрения, диплопией, расстройством глотания, снижением слуха по типу кохлеарного неврита, системным головокружением с иллюзией вращения окружающих предметов, ушным шумом, синдромом сонных апноэ, повторяющимися кратковременными потерями сознания, ортостатическим коллапсом. Пациенты, у которых имеется аномалия Киари, отмечают усиление головокружения и ушного шума при поворотах головой. Поворот головы у таких больных может спровоцировать обморок. Может отмечаться атрофические изменения половины языка и парез гортани, сопровождающийся осиплостью голоса и затруднением дыхания. Возможен тетрапарез с большим снижением мышечной силы в верхних конечностях, чем в нижних.
В случаях, когда аномалия Киари I сочетается с сирингомиелией, наблюдается сирингомиелический синдром: нарушения чувствительности по диссоциированному типу, онемения, мышечные гипотрофии, тазовые нарушения, нейроартропатии, исчезновение брюшных рефлексов. При этом некоторые авторы указывают на несоответствие размера и местонахождения сирингомиелической кисты распространенности расстройств чувствительности, степени выраженности парезов и мышечной гипотрофии.
Аномалия Киари II и Киари III имеют сходные клинические проявления, которые становятся заметны с первых минут жизни ребенка. Аномалия Киари II сопровождается шумным дыханием (врожденный стридор), периодами кратковременной остановки дыхания, двусторонним нейропатическим парезом гортани, нарушением глотания с забросом жидкой пищи в нос. У новорожденных аномалия Киари II проявляется также нистагмом, повышением мышечного тонуса в верхних конечностях, цианозом кожных покровов, возникающим во время кормления. Двигательные расстройства могут быть выражены в различной степени и прогрессировать вплоть до тетраплегии. Аномалия Киари III имеет более тяжелое течение и зачастую является не совместимым с жизнью нарушением развития плода.
Диагностика аномалии Киари
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. е. гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение аномалии Киари
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр.) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз аномалии Киари
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Источник