
Лимфопролиферативный синдром по мкб 10

Содержание
- Описание
- Дополнительные факты
- Причины
- Классификация
- Симптомы
- Диагностика
- Лечение
- Прогноз
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Аутоиммунный лимфопролиферативный синдром.
Аутоиммунный лимфопролиферативный синдром
Описание
Аутоиммунный лимфопролиферативный синдром. Группа генетически обусловленных заболеваний, которые возникают по причине наследственных или соматических мутаций в генах, отвечающих за различные этапы FAS – обусловленного апоптоза. Симптоматика может быть вариабельной и наиболее часто включает в себя лимфаденопатию, спленомегалию и разнообразные аутоиммунные поражения системы крови, печени, щитовидной железы. Диагностика аутоиммунного лимфопролиферативного синдрома производится на основании результатов общего и биохимического анализов крови, биопсии лимфатических узлов, генетических исследований. Специфического лечения заболевания в настоящий момент нет, применяют комбинации иммунносупрессивной и цитотоксической терапии.
Дополнительные факты
Аутоиммунный лимфопролиферативный синдром (АЛС, ALPS, синдром Канале-Смит) – группа иммунодефицитных состояний, характеризующихся аутоиммунными цитопениями, лимфаденопатией, спленомегалией. Первые данные о заболевании стали поступать в 1968-м году, после чего вскоре началось бурное изучение патологии. Изначально АЛС был отнесен к первичным иммунодефицитам, однако со временем были обнаружены формы синдрома, обусловленные соматическими мутациями в детском и подростковом организме. Данные о встречаемости у разных исследователей довольно сильно различаются, на сегодняшний момент описано более 500 случаев различных форм аутоиммунного лимфопролиферативного синдрома. Наследственные формы заболевания передаются по аутосомно-доминантному типу, при этом в развитии врожденных форм также довольно велика роль спонтанных мутаций. Среди больных с одинаковой частотой встречаются как мальчики, так и девочки.
Аутоиммунный лимфопролиферативный синдром
Причины
Выяснено, что причиной любого типа АЛС является нарушение FAS-опосредованного апоптоза лимфоцитов. При образовании Т-лимфоцитов те линии, которые способны атаковать собственные ткани, уничтожаются за счет активизации рецепторов CD-95 (Fas-рецепторов) на поверхности их мембраны. Активация CD-95, относящегося к группе рецепторов фактора некроза опухолей, запускает многостадийную реакцию с участием каспаз, которая оканчивается апоптозом клетки. При аутоиммунном лимфопролиферативном синдроме генетические мутации приводят к блоку этого процесса на определенном этапе, из-за чего устранения потенциально опасных клонов Т-лимфоцитов не происходит, и они начинают накапливаться в лимфатических узлах. Кроме того, создаются условия для аутоиммунного поражения органов и тканей.
Наиболее часто встречаются наследственные и спонтанные мутации в гене TNFRSF6, который кодирует собственно Fas-рецептор. При этом нарушение структуры белка (особенно домена, отвечающего за взаимодействие с FADD-молекулой) приводит к тому, что он становится неспособным выполнять свои рецепторные функции и активизировать апоптоз. Возможны и соматические мутации в гене FAS, которые в полной мере проявляют себя в позднем детском или подростковом периоде, и поэтому их относят к отдельной группе АЛС. Второй по распространенности вариант аутоиммунного лимфопролиферативного синдрома обусловлен мутацией в гене CASP10, кодирующем цистин-аспарагин кислотную протеазу (каспаза-10). Этот белок играет ключевую роль в передаче сигнала об апоптозе с клеточной мембраны в ядро клетки. К этому же варианту относят и мутации гена CASP8.
Третьим по распространенности является аутоиммунный лимфопролиферативный синдром, который вызван мутацией в гене FASLG, кодирующем Fas-лиганд или рецептор CD-178. Он играет вспомогательную роль в распознавании факторов, стимулирующих апоптоз, и участвует в передаче сигнала в клетку. Некоторые формы АЛС обусловлены мутацией гена NRAS, который кодирует «малый G-белок», принимающий участие в качестве вторичного мессенджера в передаче сигналов с мембраны в клетку, в том числе и ядро. Примерно в трети случаев аутоиммунного лимфопролиферативного синдрома врачам-иммунологам не удается установить непосредственную причину заболевания.
Классификация
При помощи методов современной генетики удалось выявить шесть основных форм АЛС:
ALPS 1A. Вызвана мутацией гена TNFRSF6, расположенного на 10 – й хромосоме, чаще всего имеет врожденный характер, наследуется по аутосомно – доминантному типу. По статистике, более 40% АЛС относятся именно к этой разновидности.
ALPS 1В. Обусловлена мутацией гена FASLG, также довольно часто приводит к врожденному аутоиммунному лимфопролиферативному синдрому. К этому типу относят около 10% от всех клинических случаев АЛС.
ALPS 1m Ее причиной являются соматические мутации в гене FAS, возникающие в детском или подростковом возрасте и поэтому приводящие к поздним формам АЛС. При этом повреждение гена должно произойти в полипотентной клетке-предшественнице, которая способна дать начало многим линиям лимфоцитов. При этой форме наиболее часто возникает внезапная самопроизвольная ремиссия заболевания.
ALPS 2. Вызвана мутацией в генах CASP10 и, по некоторым данным, CASP8, которые кодируют белки – каспазы, передающие сигнал об апоптозе от рецептора к ядру клетки. Эта форма аутоиммунного лифопролиферативного синдрома составляет примерно 25% от всех случаев заболевания, может быть как врожденной, так и проявиться в более старшем возрасте.
ALPS 3. Мутация какого гена и характер ее наследования при этой форме неизвестны. Особенностью такого варианта АЛС является нарушение не только FAS-, но и IL2-опосредованного апоптоза, а также более тяжелый характер течения.
ALPS 4. Обусловлена мутацией гена NRAS, также кодирующего белки – передатчики внутриклеточного сигнала. Данный тип аутоиммунного лимфопролиферативного синдрома характеризуется более доброкачественным течением и умеренной выраженностью симптомов.
Симптомы
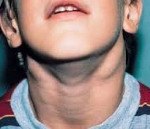
Симптомы АЛС довольно вариабельны из-за большого количества мутаций, которые могут приводить к такому состоянию. Начало заболевания можно заметить уже на 15-й день после рождения (при врожденных формах), в детском или подростковом возрасте в случае соматических мутаций в генах FAS, CASP10 или NRAS. Обычно первым проявлением заболевания является лимфаденопатия – подмышечные, паховые или шейные лимфатические узлы увеличиваются в размерах, но при этом безболезненны и не спаяны с окружающими тканями. Регистрируется спленомегалия, в некоторых случаях она сопровождается увеличением печени (гепатоспленомегалия).
Аутоиммунные проявления АЛС регистрируются обычно через некоторое время после лимфаденопатии и увеличения селезенки. В основном это поражения кровяных ростков – тромбоцитопения, гемолитическая анемия, приводящая к желтухе, изредка нейтропения. Помимо крови, аутоиммунному поражению могут подвергаться органы ЖКТ (возникают гастрит, панкреатит, колит, аутоиммунный гепатит). На коже могут проявляться признаки васкулита, делая клинику аутоиммунного лимфопролиферативного синдрома схожей с таковой при системной красной волчанке. Кроме того, могут возникать аутоиммунные формы тиреоидита, гломерулонефрита, поражаться суставы, ткани глаза (иридоциклит, увеит). Нередки поражения центральной нервной системы – эпилептические припадки, миелиты, мозжечковая атаксия.
Увеличение паховых лимфоузлов. Увеличение подмышечных лимфоузлов. Увеличение шейных лимфоузлов. Эозинофилия.
Диагностика
Диагностика АЛС производится на основании осмотра, а также лабораторных, иммунологических и генетических исследований. При осмотре выявляют увеличение более чем трех групп лимфатических узлов, спленомегалию, увеличение печени. Анализ крови может показывать уменьшение количества некоторых клеток (анемию, тромбоцитопению), у части больных определяется высокая (до 30%) эозинофилия. Проба Кумбса положительная, в биохимическом анализе крови определяется выраженная гипергаммаглобулинемия. Одним из высокочувствительных методов иммунологической диагностики аутоиммунного лимфопролиферативного синдрома является проточная иммуноцитофлюориметрия, проводимая с целью выявления количества лимфоцитов с атипичным набором рецепторов (CD3+CD4-CD8-). При АЛС количество таких клеток превышает 1% от всех лимфоцитов. В биоптате лимфатических узлов определяется фолликулярная гиперплазия, результатом гистологического исследования селезенки служит лимфоидная гиперплазия.
Врачом-генетиком может быть произведено секвенирование гена FAS с целью выявления мутаций, ставших причиной аутоиммунного лимфопролиферативного синдрома. С учетом значительной величины этого гена для ускорения и удешевления процедуры поиск может быть произведен лишь в отдельных экзонах гена FAS, в которых наиболее часто обнаруживаются нарушения – эти участки называют «горячими точками». Таким образом, при помощи генетической диагностики можно определить АЛС только 1А, 1В и 1m типов. Методики определения остальных форм АЛС генетическими методами на сегодняшний день не разработаны. Изучение наследственного анамнеза в ряде случаев будет неэффективно из-за значительной доли форм заболевания, вызванных соматическими мутациями.
Лечение
Этиотропное лечение аутоиммунного лимфопролиферативного синдрома не разработано, патогенетическая терапия сводится к применению иммуносупрессивных и цитотоксических средств. В качестве средств, подавляющих аутоиммунную активность, наиболее часто используют кортикостероиды (преднизолон, дексаметазон). К специфическим препаратам, ограничивающим скорость пролиферации лимфоцитов, относят микофенолата мофетил, сиролимус. Также при аутоиммунном лимфопролиферативном синдроме активно применяются традиционные цитотоксические средства – метотрексат, циклоспорин А и другие. При значительном увеличении селезенки или отсутствии эффекта от консервативного лечения прибегают к спленэктомии. Пересадка костного мозга и использование стволовых клеток в долгосрочной перспективе давали только временный эффект. При значительно выраженных гематологических нарушениях применяют гемотрансфузии, введение эритроцитарной или тромбоцитарной массы. Больному следует избегать физических нагрузок, использовать высоковитаминную диету.
Прогноз
Прогноз заболевания, ввиду высокой вариабельности и выраженности симптомов, неопределенный или неблагоприятный. У большей части больных проявления заболевания постепенно нарастают, со временем приводя к летальной анемии, тромбоцитопении, билиарному циррозу печени. Также важную роль в прогнозе играют нарушения иммунитета, так как нередко причиной смерти выступают сепсис и другие инфекционные поражения. В прогнозе аутоиммунного лимфопролиферативного синдрома следует учитывать и повышенный риск онкологических заболеваний, примерно пятая часть больных умирает от различных типов лимфом. В некоторых случаях возникает спонтанная и длительная ремиссия патологии.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Медицинский эксперт статьи

х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Аутоиммунный лимфопролиферативный синдром (АЛПС) – заболевание, в основе развития которого лежат врожденные дефекты Fas-опосредоаанного апоптоза. Оно было описано в 1995 г., но еще с 60-х годов заболевание со схожим фенотипом было известно под названием синдром CanaLe-Smith.
Заболевание характеризуется хронической незлокачественной лимфопролиферацией и гипергаммаглобулинемией, которые могут сочетаться с различными аутоиммунными нарушениями.
 [1], [2], [3], [4], [5]
[1], [2], [3], [4], [5]
Код по МКБ-10
D72.8 Другие уточненные нарушения белых кровяных телец
D89.8 Другие уточненные нарушения с вовлечением иммунного механизма, не классифицированные в других рубриках
D82.8 Иммунодефицит, связанный с другими уточненными значительными дефектами
Патогенез
Апоптоз, или физиологическая гибель клетки, является одним из неотъемлемых механизмов поддержания гомеостаза организма. Апоптоз развивается вследствие активации различных сигнальных механизмов. Особую роль в регуляции системы гемопоэза и иммунной системы играет апоптоз, опосредованный активацией Fas-рецепторов (CD95) при их взаимодействии с соответствующим лигандом (Fas-лиганд, FasL). Fas представлен на различных гемопоэтических клетках, высокая экспрессия Fas рецептора характерна для активированных лимфоцитов. Fasl-экспрессируется, главным образом, CD8+T-лимфоцитами.
Активация Fas рецептора влечет за собой ряд последовательных внутриклеточных процессов, итогом которых является дезорганизация ядра клетки, денатурация ДНК, изменения мембраны клетки, приводящие к ее распаду на ряд фрагментов без выброса во внеклеточную среду лизосомальных ферментов и без индукции воспаления. В передаче апоптотического сигнала к ядру участвует ряд ферментов, называемых каспазами, в том числе каспаза 8 и каспаза 10.
Fas-опосредованный апоптоз играет важную роль в элиминации клеток с соматическими мутациями, аутореамтивных лимфоцитов, а также лимфоцитов, выполнивших свою роль в процессе нормального иммунного ответа. Нарушение апоптоза Т-лимфоцитов приводит к экспансии активированных Т-клеток, а также так называемых двойных негативных Т-лимфоцитов, которые экспрессируют Т-клеточный рецептор с a/b цепями (TCRa/b), но не имеют ни CD4, ни CD8 молекул. Дефект программируемой гибели В-клеток в совокупности с повышением уровня интерлейкина 10 (IL-10) приводят к гипергаммаглобулинемии и повышению выживаемости аутореактивных В-лимфоцитов. Клиническими последствиями являются избыточное накопление лимфоцитов в крови и лимфоидных органах, увеличение риска аутоиммунных реакций и опухолевого роста.
К настоящему времени выявлено несколько молекулярных дефектов, приводящих к нарушению апоптоза и развитию АЛЛС. Это мутации в генах Fas, FasL, каспазы 8 и каспаэы 10.
[6], [7], [8], [9], [10], [11], [12], [13]
Симптомы аутоиммунного лимфопролиферативного синдрома
АЛПС отличается большой вариабельностью спектра клинических проявлений и тяжести течения, и возраст клинической манифестации также может колебаться в зависимости от выраженности симптоматики. Известны случаи дебюта аутоиммунных проявлений во взрослом возрасте, когда и был диагностирован АЛПС. Проявления лимфопролиферативного синдрома присутствуют с рождения в виде увеличения всех групп лимфоузлов (периферических, внутригрудных, внутрибрюшных), увеличения размеров селезенки, а часто и печени. Размеры лимфоидных органов могут изменяться в течение жизни, иногда отмечено их нарастание при интеркуррентных инфекциях. Лимфоузлы имеют обычную консистенцию, иногда плотноваты; безболезненны. Известны случаи резко выраженных проявлений гиперпластического синдрома, имитирующих лимфому, с увеличением периферических лимфоузлов, приводящим к деформации шеи, гиперплазией внутригрудных лимфоузлов вплоть до развития синдрома сдавления и дыхательной недостаточности. Описаны лимфоидные инфильтраты в легких. Однако во многих случаях проявления гиперпластического синдрома не столь драматичны, и они остаются незамеченными врачами и родителями. Степень выраженности спленомегалии также весьма вариабельна.
Тяжесть течения заболевания определяется главным образом аутоиммунными проявлениями, которые могут развиться в любом возрасте. Чаще всего встречаются различные иммунные гемопатии – нейтропения, тромбоцитопения, гемолитическая анемия, которые могут сочетаться в виде двух- и трехростковых цитопений. Может иметь место единичный эпизод иммунной цитопении, но зачастую они носят хронический или рецидивирующий характер.
Из других, более редких аутоиммунных проявлений, могут наблюдаться аутоиммунный гепатит, артрит, сиаладенит, воспалительные заболевания кишечника, узловатая эритема, панникулит, увеит, синдром Guiltain-Barre. Кроме того, могут наблюдаться различные кожные сыпи, преимущественно уртикарные, субфебрилитет или лихорадка без связи с инфекционным процессом.
У больных аутоиммунным лимфопролиферативным синдромом увеличена частота развития злокачественных опухолей по сравнению с популяцией. Описаны случаи гемобластозов, лимфом и солидных опухолей (карцинома печени, желудка).
[14]
Формы
В 1999 г. предложена рабочая классификация аутоиммунного лимфопролиферативного синдрома, основанная на типе дефекта апоптоза:
- ALP5 0 – полный дефицит CD95, являющийся следствием гомозиготной нуль-мутации (homozygous nuLl mutation) в гене Fas/CD95;
- ALPS I – дефект передачи сигнала через Fas-рецептор.
- При этом ALPS la является следствием дефекта Fas-рецептора (гетерозиготная мутация в гене Fas);
- ALPS lb является следствием дефекта Fas-лиганда (FasL), связанного с мутацией в соответствующем гене – FASLG/CD178;
- ALPS Ic является следствием только что выявленной гомозиготной мутации в гене FA5LG/CD178;
- ALPS II – дефект внутриклеточной передачи сигнала (мутация в гене каспазы 10 – ALPS IIа, в гене каспазы 8 – ALPS IIb);
- ALPS III – молекулярный дефект не установлен.
Тип наследования
АЛПС 0 типа – полный дефицит CD95 – описан всего лишь у нескольких пациентов, Поскольку гетерозиготные члены семей не имеют фенотипа АЛПС, была предложена гипотеза о аутосомно-рецессивном типе наследования. Однако, неопубликованные данные о наблюдении за семьей, в которой был выявлен пациент с АЛПС 0, не полностью согласуются с данным утверждением. Ученые выяснили, что многие, если не все, мутации являются доминантными, и что если они оказываются гомозиготными, это приводит к более выраженному фенотипу заболевания.
При АЛПС I типа тип наследования – аутосомно-доминантный, с неполной пенетрантностью и вариабельной экспрессивностью. В частности, при АЛПС1а описаны случаи гомозиготности или сочетанной гетерозиготности, при которой определяются различные мутации гена Fas в обеих аллелях. Эти случаи характеризовались тяжелым течением с пренатальной или неонатальной манифестацией (водянка плода, гепатоспленомегалия, анемия, тромбоцитопения). Кроме того, выявлена корреляция выраженности клинической симптоматики с типом мутации в гене Fas; для мутации во внутриклеточном домене характерно более тяжелое течение. Всего в мире описано более 70 пациентов с ALPS la. Мутация FasL была впервые описана у пациента с клиническими проявлениями системной красной волчанки и хронической лимфопролиферацией. Она была категоризирована как ALPS lb, хотя фенотип не полностью отвечал критериям классического аутоиммунного лимфопролиферативного синдрома (двойные негативные Т-клетки и спленомегалия отсутствовали). Первая гомозиготная мутация А247Е в гене FasL (внеклеточный домен) была описана недавно, в 2006 году, Del-Rey M et al. у пациента с нелетальным АЛПС, что говорит о важной роли терминального домена FasL C0OH во взаимодействии Fas/FasL. Авторы предлагают в действующую классификацию аутоиммунного лимфопролиферативного синдрома внести подгруппу ALPS Ic.
АЛПС II типа наследуется по аутосомно-рецессивному типу, и у многих пациентов с этим типом заболевания отмечалась типичная клиническая и иммунологическая АЛПС, включая нарушенный Fas-опосредованный апоптоз, в реализации которого задействованы как каспаза 8 (вовлечена на ранних этапах межклеточной передачи сигнала на уровне взаимодействий TCR и BCR), так и каспаза 10 (вовлечена в апоптотический каскад на уровне всех известных рецепторов, которые индуцируют апоптоз лимфоцитов).
Более чем у 30 пациентов была выявлена клиническая картина АЛПС средней степени выраженности, включавшая в себя гипергаммаглобулинемию и повышенный уровень двойных негативных Т-клеток в крови, причем активированные лимфоциты пациентов с АЛПС III типа (именно так был назван этот синдром) показывали нормальную активацию Fas-опосредованного пути in vitro, и никаких молекулярных дефектов найдено не было. Возможно, причиной заболевания служат нарушений других апоптотических путей, опосредованных, например, Trail-R, DR3 или DR6. Интересным кажется наблюдение R. Qementi о выявлении мутации N252S в гене перфорина (PRF1) у больного с АЛПС III типа, у которого наблюдалось существенное снижение НК-активности. При этом автор отмечает, что значительная разница между частотой обнаружения N252S у пациентов с АЛПС (2 из 25) и частотой ее выявления в группе контроля (1 из 330) заставляет предполагать ее связь с развитием АЛПС в итальянской популяции. С другой стороны, F. Rieux-Laucat отмечает, что данный вариант мутации PRF1 выявлялся им у 18% здоровых и у 10% больных АЛПС (неопубликованные данные). И, кроме того, наряду с полиморфизмом N252S, им найдена мутация гена Fas у пациента с АЛПС и его здорового отца, что, по мнению F.Rieux-Laucat, говорит о непатогенности гетерозиготной мутации N252S в гене перфорина, описанной несколько ранее R. Qementi у пациента с АЛПС (мутация Fas) и крупноклеточной В-лимфомой. Таким образом, вопрос о причинах возникновения АЛПС III типа на сегодняшний день остается открытым.
[15], [16], [17], [18]
Диагностика аутоиммунного лимфопролиферативного синдрома
Одним из признаков лимфопролиферативного синдрома может быть абсолютный лимфоцитоэ в периферической крови и костном мозге. Содержание лимфоцитов повышается за счет В- и Т-лимфоцитов, в некоторых случаях – только за счет одной из субпопуляций,
Характерно увеличение содержания в периферической крови двойных негативных лимфоцитов с фенотипом CD3+CD4-CD8-TCRa/b. Эти же клетки находят в костном мозге, лимфоузлах, лимфоцитарных инфильтратах в органах.
Снижение экспрессии CD95 (Fas-рецептора) на лимфоцитах не является диагностическим критерием аутоиммунного лимфопролиферативного синдрома, поскольку его уровень может оставаться в пределах нормы при некоторых дефектах Fas с мутацией во внутриклеточном домене, а также при АЛПС II и III типа.
Типичным признаком аутоиммунного лимфопролиферативного синдрома является гипериммуноглобулинемия, причем за счет повышения уровня как всех, так и отдельных классов иммуноглобулинов. Степень повышения может быть различной.
Известны единичные случаи аутоиммунного лимфопролиферативного синдрома с гипоиммуноглобулинемией, природа, которой не выяснена. Иммунодефицит более характерен для больных с АЛПС IIb, хотя описан и при АЛПС 1а типа.
У больных могут обнаруживаться различные аутоантитела: антитела к клеткам крови, АНФ, антитела к нативной ДНК, анти-RNP, анти-SM, анти-SSB, РФ, антитела в VIII фактору свертывания.
Сообщают о повышенном содержании триглицеридов в сыворотке больных аутоиммунным лимфопролиферативным синдромом; предполагается вторичный характер гипертриглицеридемии вследствие повышенной продукции цитокинов, влияющих на липидный обмен, в частности, фактора некроза опухоли (tumor necrosis factor, TNF). Значительное повышение содержания TNF обнаруживается у большинства больные аутоиммунным лимфопролиферативным синдромом. У некоторых пациентов уровень гипертриглицеридемии коррелирует с течением заболевания, повышаясь при обострениях.
Необходимость дифференциальной диагностики со злокачественными лимфомами обусловливает показания к проведению открытой биопсии лимфатического узла. Морфологическое и иммуногистохимическое исследование лимфатического узла обнаруживает гиперплазию паракортикальных зон и, в ряде случаев, фолликулов, инфильтрацию Т- и В-лимфоцитами, иммунобластами, плазматическими клетками. В отдельных случаях находят гистиоциты. Структура лимфоузла, как правило, сохранена, в отдельных случаях может быть несколько стерта за счет выраженной смешанной клеточной инфильтрации.
У больных, подвергшихся спленэктомию по поводу хронический иммунных гемопатий, выявляют смешанную лимфоидную инфильтрацию, в том числе, клетками двойной негативной популяции.
Специфическим методом диагностики аутоиммунного лимфопролиферативного синдрома является исследование апоптоза периферических мононуклеаров (ПМН) больного in vitro, при индукции моноклональными антителами к Fas-рецептору. При АЛПС отсутствует увеличение числа апоптотических клеток при инкубации ПМН с анти-FasR антителами.
Молекулярно-диагностические методы направлены на выявление мутации в генах Fas, каспазы 8 и каспазы 10. В случае нормальных результатов апоптоза ПМН и наличии фенотипической картины АЛПС показано исследование гена FasL
[19], [20], [21]
Какие анализы необходимы?
Дифференциальная диагностика
Дифференциальный диагноз аутоиммунного лимфопролиферативного синдрома проводят со следующими болезнями:
- Инфекционные заболевания {вирусные инфекции, туберкулез, лейшманиоз и др.)
- Злокачественные лимфомы.
- Гемофагоцитарный лимфогистиоцитоз.
- Болезни накопления (болезнь Гоше).
- Саркоидоз.
- Лимфаденопатия при системных завоеваниях соединительной ткани.
- Другие иммунодефицитные состояния (общая вариабельная иммунная недостаточность, синдром Вискотта-Олдрича).
Лечение аутоиммунного лимфопролиферативного синдрома
При изолированном лимфопролиферативном синдроме терапии, как правило, не требуется, за исключением случаев резко выраженной гиперплазии с синдромом сдавления средостения, развитием лимфоидных инфильтратов в органах. При этом применяют иммуносупрессивную терапию (глюкокортикоиды, циклоспорин А, циклофосфамид),
Лечение аутоиммунных осложнений проводят по общим принципам терапии соответствующих заболеваний – при гемопатиях назначают (метил)преднизолон в дозе 1-2 мг/кг, либо в режиме пульс-терапии с последующим переходом на поддерживающие дозы; при недостаточном или неустойчивом эффекте применяют комбинацию кортикостероидов с другими иммуносупрессорами, например: мофетила микофенолат, циклоспорин А, азатиоприн, моноклональными антителами к анти-СD20 (ритуксимаб). Терапия высокими дозами внутривенного иммуноглобулина (ВВИГ), как правило, дает неудовлетворительный или нестойкий эффект. В связи со склонностью к хроническому или рецидивирующему течению, необходима длительная терапия поддерживающими дозами, которые подбирают индивидуально. При недостаточном эффекте медикаментозной терапии, потребности в высоких дозах препаратов, может оказаться эффективной спленэктомия.
В случае тяжелого течения или прогнозируемой прогрессии заболевания показано проведение трансплантации гемопоэтических стволовых клеток, однако опыт проведения трансплантаций при аутоиммунном лимфопролиферативном синдроме ограничен во всем мире.
Прогноз
Прогноз зависит от тяжести течения заболевания, которая чаще всего определяется выраженностью аутоиммунных проявлений. При тяжелых, резистентных к терапии гемопатиях, вероятен неблагоприятный исход.
С возрастом возможно уменьшение выраженности лимфопролиферативного синдрома, однако это не исключает риск манифестации тяжелых аутоиммунных осложнений. В любом случае, адекватный прогноз помогает выработать оптимальный терапевтический подход к каждому пациенту.
[22], [23], [24]
Источник