
Лиссэнцефалия код по мкб 10

Содержание
- Описание
- Дополнительные факты
- Симптомы
- Причины
- Классификация
- Диагностика
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Название: Лиссэнцефалия.
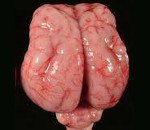
Лиссэнцефалия
Описание
Лиссэнцефалия. Группа генетически обусловленных аномалий развития головного мозга, характеризующихся частичным или полным недоразвитием извилин и борозд коры больших полушарий, а также нарушением ее ультраструктуры. Выраженность и сочетания симптомов этого состояния различаются при разных формах заболевания, наиболее распространены судороги, глубокая умственная отсталость, нарушения глотания и гипотония мышц. Диагностика лиссэнцефалии может производиться ультразвуковыми методиками (в том числе и пренатально), компьютерной и магнитно-резонансной томографией, для наиболее распространенных форм возможно определение посредством молекулярно-генетического анализа. Специфического лечения не существует, используют симптоматическую и поддерживающую терапию.
Дополнительные факты
Лиссэнцефалия – группа тяжелых аномалий развития головного мозга, которые сопровождаются недоразвитием коры больших полушарий с формированием пахигирии (наличием всего нескольких извилин и борозд) или агирии (полным отсутствием складчатости коры). Данная патология может выступать в качестве самостоятельного генетического заболевания или входить в симптомокомплекс других синдромов – например, Миллера-Дикера, Фукуямы и Уокера-Варбурга. Механизм наследования различных типов лиссэнцефалии может быть аутосомно-рецессивным, аутосомно-доминантным (в данном случае чаще всего имеют место спонтанные или герминативные мутации) и сцепленным с Х-хромосомой. Из-за многообразия механизмов наследования половое распределение нарушения неодинаково при различных формах патологии. Лиссэнцефалия является достаточно редкой генетической аномалией развития мозга, поэтому встречаемость определена только для наиболее распространенной первой группы нарушений – она составляет 11,7 случаев на 1 000 000 новорожденных. Для остальных групп лиссэнцефалии встречаемость не установлена, в том числе и потому, что во многих случаях плод с подобной патологией не вынашивается, и беременность самопроизвольно прерывается в первом триместре еще до определения наличия порока.
Лиссэнцефалия
Симптомы
Судороги.
Причины
Основная общая причина всех типов лиссэнцефалии – нарушение процесса миграции клеток-предшественников нейронов (нейробластов) из передних отделов нервной трубки к будущей коре больших полушарий. В результате этого вместо сложной складчатой структуры, которая имеет в своем составе шесть слоев, образуется гладкая или имеющая в разы меньше борозд кора, состоящая из 2-4 слоев (в зависимости от формы заболевания). Поскольку кора больших полушарий у человека отвечает за когнитивные функции, в ней содержится огромное число нервных центров и обширные ассоциативные зоны, лиссэнцефалия приводит к тяжелейшим расстройствам. Кроме того, при некоторых типах мутаций, вызывающих данное состояние, возможно развитие аномалий других органов и тканей, что еще больше усугубляет состояние больного.
Наиболее распространенные типы лиссэнцефалии обусловлены дефектами гена PAFAH1B1, также известного под названием LIS1 и расположенного на 17-й хромосоме. Многие врачи-генетики отмечают, что для возникновения выраженного недоразвития коры головного мозга и синдрома Миллера-Дикера необходимы не точечные мутации в LIS1, а крупные делеции с захватом сотен пар азотистых оснований. Нередко при этом повреждаются и окружающие гены, что становится причиной разнообразных фенотипических проявлений данного типа лиссэнцефалии. Ген LIS1 кодирует внутриклеточную субъединицу сложного по своей структуре фермента, который принимает активное участие в миграции нейробластов. Дефекты в структуре этой субъединицы приводят к нарушению данного процесса и развитию данного заболевания.
Другая форма лиссэнцефалии обусловлена мутацией гена DCX, локализованного на Х-хромосоме, поэтому наследование этого типа патологии сцеплено с полом. Белок, получаемый в результате экспрессии данного гена, участвует в формировании особого типа микротрубочек, которые вырабатываются только нейробластами и необходимы им для формирования связей между клетками. Нарушения в структуре DCX приводят к синтезу дефектного белка, что провоцирует лиссэнцефалию. Другой относительно изученный вариант этого состояния, также сцепленный с Х-хромосомой, вызывается мутациями гена ARX, который является фактором транскрипции для иных генов. Он контролирует развитие коры больших полушарий, поджелудочной железы и половых органов, из-за чего дефекты ARX проявляются многочисленными пороками данных органов, в том числе лиссэнцефалией.
Еще одним распространенным вариантом лиссэнцефалии является форма заболевания, обусловленная мутацией гена RELN, расположенного на 7-й хромосоме. Дефекты этого гена приводят к так называемому синдрому Норман-Робертса, который, помимо прочего, сопровождается выраженным нарушением складчатости коры больших полушарий. Ген RELN кодирует последовательность гликопротеида рилина, который принимает активное участие в формировании нервной ткани, а также контролирует образование новых дендритов и функционирование долговременной памяти у взрослых людей. Удалось идентифицировать еще один ген, приводящий к развитию лиссэнцефалии – TUBA1A, который локализован на 12-й хромосоме. Он кодирует определенный компонент цитоскелета нейронов, его дефект приводит к неполноценности нейробластов, что нарушает процесс их миграции в эмбриональном периоде.
Классификация
На сегодняшний день выявлено более двадцати вариантов лиссэнцефалии. Эти варианты обусловлены различными мутациями, имеют отличия в фенотипических проявлениях заболевания, разные механизмы наследования и тяжесть симптомов. Долгое время разновидности патологии не удавалось успешно классифицировать из-за значительного разнообразия генетических дефектов и недостаточных данных о ключевых генах, приводящих к развитию некоторых форм заболевания. Лишь в 2003-м году удалось создать достаточно приемлемую с точки зрения современной генетики и неврологии классификацию лиссэнцефалии, которая учитывает основные нюансы этого состояния. Специалисты разделяют все формы лиссэнцефалии на пять классов:
• Класс 1, часто называемый классической лиссэнцефалией. Включает в себя формы заболевания, обусловленные мутациями гена LIS1 (изолированный тип и синдром Миллера-Дикера), а также сцепленную с полом разновидность, вызванную мутацией DCX. Кроме того, в этот класс часто включают некоторые типы лиссэнцефалии с неясными генетическими причинами. Особенностью группы является нарушение складчатости коры больших полушарий с минимальным проявлением иных аномалий центральной нервной системы.
• Класс 2. В настоящее время состоит только из одного типа лиссэнцефалии, которая вызывается дефектами гена ARX. Заболевание сцеплено с Х-хромосомой, помимо пороков развития коры у больных часто обнаруживаются отсутствие мозолистого тела, нарушения терморегуляции, выраженные аномалии половых органов и поджелудочной железы.
• Класс 3. Включает в себя разновидности лиссэнцефалии в сочетании с гипоплазией или полным недоразвитием мозжечка, наиболее типичной мутацией для данного класса являются дефекты гена RELN. Может регистрироваться как изолированный вариант патологии или в составе синдрома Норман-Робертса.
• Класс 4, нередко называется микролиссэнцефалией, поскольку нарушение формирования коры больших полушарий сопровождается выраженной микроцефалией. Некоторые формы этого заболевания были достоверно ассоциированы с мутациями гена TUBA1A.
• Класс 5 или булыжниковые лиссэнцефалии, к которым относят синдромы Фукуямы и Уокера-Варбурга. Последний соотносится с дефектами генов POMT1, POMT2, FKRP и некоторыми другими, все они расположены на 9-й хромосоме.
Данная классификация критикуется некоторыми исследователями по причине наличия большого количества «белых пятен» в виде включения в нее форм лиссэнцефалии с неопределенными ключевыми генами. Однако в настоящее время именно это разделение считается наиболее общепринятым в научном мире. Классификация корректируется по мере дальнейшего изучения данного состояния.
Диагностика
В отношении лиссэнцефалии возможна пренатальная диагностика при помощи ультразвуковых методов исследования, при этом увеличение разрешающей способности УЗИ-аппаратуры способствует все более раннему определению заболевания. Развитие нарушений в строении коры больших полушарий происходит на 14-20-й неделе гестации, в настоящее время уже в этот период можно определить патологию и поставить вопрос о прерывании беременности. После рождения ребенка с подозрением на лиссэнцефалию диагноз подтверждают при помощи КТ и МРТ. Молекулярно-генетическая диагностика обладает высокой точностью, но она доступна только в отношении тех форм заболевания, для которых известны ключевые гены. Специфического лечения лиссэнцефалии не существует, применяют противосудорожные препараты, ноотропы и другие средства, позволяющие уменьшить выраженность симптоматики. При наличии иных пороков развития производится их коррекция по медицинским показаниям.
Лечение
Прогноз практически любой формы лиссэнцефалии крайне неблагоприятный, большинство больных умирает в раннем детстве от осложнений и других пороков развития. Описаны отдельные легкие случаи этого состояния с частичным или очаговым недоразвитием коры больших полушарий, но не все специалисты склонны причислять такие типы патологии к лиссэнцефалиям. Профилактика возможна только в рамках пренатальной диагностики. При наличии в роду подобных заболеваний имеет смысл провести генетический анализ на носительство генов аутосомно-рецессивных типов патологии. При обнаружении у плода нарушений формирования мозга, соответствующих лиссэнцефалии, ставится вопрос о прерывании беременности.
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник
Лиссэнцефалия (в переводе с др.-греч. «гладкий головной мозг») – аномалия развития, для которой характерно сглаживание извилин коры больших полушарий головного мозга.
Возникает вследствие недостаточной миграции зародышевых нервных клеток (нейробластов) из первичной нервной трубки.
Наиболее тяжелая форма заболевания – агирия, при которой извилины мозга вообще отсутствуют.
Общие сведения
Лиссэнцефалия – редко встречающийся порок развития, который может быть как самостоятельной аномалией развития, так и проявлением некоторых врожденных синдромов (Миллера-Дикера и Нормана-Робертса). Наблюдается у детей независимо от пола. Распространенность заболевания в настоящее время не установлена.
Формы
Лиссэнцефалия может быть тотальной и очаговой.
В зависимости от тяжести нарушений выделяют:
- агирию, при которой поверхность мозга лишена извилин;
- пахигирию, которая отличается наличием нескольких плоских широких извилин и неглубоких борозд.
В зависимости от морфологических и генетических данных с 2003 года выделяется пять групп данной аномалии.
Первая группа — классический тип заболевания (I тип лиссэнцефалии в предшествующей классификации), которая включает:
- лиссэнцефалию, связанную с мутацией гена LIS1;
- лиссэнцефалию, которая вызывается мутацией гена DCX (doublecortin);
- изолированную лиссэнцефалию I типа;
- синдром Миллера-Дикера;
- изолированную лиссэнцефалию I типа, которая возникла по неустановленным генетическим причинам.
Во вторую группу включена форма, которая связана с X-сцепленным рецессивным наследованием и сопровождается врожденным отсутствием мозолистого тела. Заболевание вызывается мутацией гена ARX.
Третья группа включает лиссэнцефалию, при которой наблюдается врожденное недоразвитие (гипоплазия мозжечка). К этой же группе относится синдром Норман-Робертс, вызванный мутацией гена RELN.
К четвертой группе относят микролиссэнцефалию.
В пятую группу входит «булыжниковая» лиссэнцефалия (II тип по старой классификации). Включает:
- синдром Уокера-Варбурга (HARD(E)-синдром);
- синдром Фукуямы;
- Мышечно-Глазо-Мозговую Болезнь (MEB).
Существует также морфологическая классификация, выделяющая:
- 1 степень, которая ставится при полной диффузной агирии без контурирующихся извилин.
- 2 степень, которая ставится при наличии «расширенной» агирии в сочетании с присутствием отдельных извилин (обычно эти извилины расположены в височных и лобных отделах).
- 3 степень — при обширных участках агирии и пахигирии. В большинстве случаев пахигирия отмечается в лобных отделах, а агирия – в теменных и центральных отделах мозга
- 4 степень ставится при «расширенной» пахигирии (участки агирии отсутствуют).
- 5 степень ставится при наличии симметричных участков пахигирии, которые перемежаются с фрагментами коры, соответствующей норме.
Причины развития
Лиссэнцефалия развивается в результате нарушения миграции нейронов при формировании и развитии коры головного мозга.
Нарушение миграции возможно в результате мутации:
- гена RELN (рилина), который расположен на хромосоме 7 в регионе 7q22 (развивается синдром Норман-Робертс);
- гена ARX, расположенного на Х-хромосоме в регионе p21.3.
К нарушениям формирования коры головного мозга приводит и частичная утрата (делеция) гена LIS-1, расположенного в хромосоме 17 регион р13.3 и принимающего участие в миграции нейронов (аутосомно-рецессивный тип наследования).
При утрате некоторых генов 17-й хромосомы в регионе 13 (PAFAH1B1) развивается синдром Миллера-Дикера.
К развитию лиссэнцефалии приводят также нарушения X-сцепленных генов. При мутациях гена XLIS у детей мужского пола диагностируют лиссэнцефалию, а у детей женского пола – двойную кору, так как в результате нарушения миграции слой серого вещества откладывается под белым веществом головного мозга (в норме должно быть наоборот). Дефектный ген кодируется белком doublecortin (DCX), поэтому его мутация тоже может стать причиной развития аномалии. Тип наследования — Х-сцепленный рецессивный.
Причиной развития «булыжниковой» лиссэнцефалии являются:
- Дефекты 9-й хромосомы в регионе q34 (синдром Уокера-Варбурга).
- Дефекты гена FCMD, расположенного в 9-й хромосоме в регионе q31 (синдром Фукуямы). Чаще всего мутация заключается в присутствии избыточных мобильных генетических элементов первого типа.
- Мутации гена POMGNT1, который расположен в 1-й хромосоме в регионе p34 (Мышечно-Глазо-Мозговая Болезнь).
Причины некоторых форм лиссэнцефалии в настоящее время не выявлены.
Кроме генетических нарушений причиной развития аномалии может быть:
- перенесенная в первом триместре вирусная инфекция матки или плода;
- плохое кровоснабжение мозга плода на начальном этапе беременности.
Патогенез
Формирование и развитие коры головного мозга включает:
- пролиферацию (деление) нервных клеток;
- миграцию нейронов из эмбрионального матрикса;
- дифференцировку коры.
При нормальном развитии плода происходит:
- оформление полушарий мозга на 11-12 неделе внутриутробного развития;
- формирование латеральной, шпорной и опоясывающей борозды на 16 неделе эмбриогенеза;
- уплощение кортикального плато и формирование первичных и центральных борозд на 20 неделе;
- формирование ольфакторных борозд на 24-26 неделе;
- образование верхних височных борозд на 28 неделе.
Созревание коры головного мозга обеспечивается миграцией нейронов, которые располагаются в мозге слоями. В процессе миграции те клетки, которые образовались позже, мигрируют дальше своих предшественниц, образуя таким образом новый слой.
Процесс миграции регулируется белками рилином и N-кадгерином.
Большая часть борозд и извилин формируются после окончания миграции нейронов, поэтому нарушение процесса миграции нейронов приводит к нарушению формирования извилин и борозд.
Симптомы
Симптомы заболевания зависят от типа лиссэнцефалии.
Классический тип заболевания проявляется:
- отставанием в росте, психоречевом и двигательном развитии;
- относительно небольшим размером головы;
- судорогами, которые могут манифестировать с первых дней жизни или развиваться после 1,5 лет;
- симптомокомплексом «вялого ребенка».
Лиссэнцефалия при синдроме Миллера-Дикера сопровождается:
- черепно-лицевой дизморфией (изменением формы лица);
- увеличенным расстоянием между парными органами;
- замедленным ростом;
- патологиями почек, сердца, ЖКТ.
При обследовании выявляется уменьшенное количество кортикальных слоев (четыре вместо шести).
При синдроме Нормана-Робертса заболевание сопровождается:
- увеличенным расстоянием между глазами;
- микроцефалией (маленький размер головы);
- наличием покатого лба;
- эпилепсией;
- отеком тканей (лимфедемой);
- задержкой умственного развития.
Обследование позволяет выявить утолщенную кору мозга, наличие агирии, значительные изменения гиппокампа, мозжечка и ствола мозга.
При синдроме Уокера-Варбурга помимо лиссэнцефалии наблюдается:
- микрофтальмия, катаракта, отслоение сетчатки;
- аномалии развития скелета и мышц;
- гидроцефалия, цефалоцеле;
- задержка умственного развития.
При синдроме Фукуямы лиссэнцефалия сочетается с:
- прогрессирующей мышечной дистрофией;
- задержкой темпов развития;
- выраженным слабоумием;
- гидроцефалией, микрополигирией, аплазией мозолистого тела, уменьшением плотности белого вещества;
- судорогами в большинстве случаев.
В отдельных случаях выявляют атрофию зрительных нервов и патологию сетчатки.
Диагностика
Для диагностики лиссэнцефалии используются:
- пренатальное обследование на 22- 28 неделе беременности при помощи ультразвукового оборудования;
- КТ, позволяющая выявить наличие сглаженности извилин, гипоплазию белого вещества, увеличение желудочков мозга, прямолинейную границу между серым и белым веществом, расширение и вертикальную ориентацию сильвиевых щелей, расширение центральной борозды и др.;
- МРТ, которая позволяет выявить тип лиссэнцефалии по характерным признакам;
- ЭЭГ, при помощи которого определяется характерный для лиссэнцефалии быстрый и высокоамплитудный ритм в задних отделах мозга;
- генетический анализ.
При пренатальной диагностике необходимо учитывать, что в начале II-го триместра достаточно гладкий мозг – норма, поэтому оценивать формирование извилин и борозд можно не раньше 20 недели.
При подозрении на наличие аномалии возможно исследование хромосом плода.
Лечение
Специфическое лечение лиссэнцефалии не разработано.
Для улучшения состояния больных применяются:
- производные вальпроевой кислоты в качестве противосудорожной терапии;
- карнитин, который используется при мышечной дистрофии;
- церебрализин, актовегин, глицин, витамины в качестве поддерживающей терапии.
В некоторых случаях возможно оперативное вмешательство, которое заключается в трансплантации стволовых клеток. Поскольку положительные результаты пока наблюдаются редко и заключаются только в некотором улучшении состояния, а риск побочных эффектов у больного ребенка высок, операции проводят не часто.
Лиссэнцефалия – заболевание с неблагоприятным прогнозом, поэтому семьям, в которых имелось данное заболевание, необходима консультация генетика.
Источник