
Механизмы незавершенного фагоцитоза синдрома ленивых лейкоцитов

Основными клиническими проявлениями
дефекта нейтрофилов являются рецидивирующие
бактериальные и грибковые инфекции с
поражением кожи и внутренних органов.
Основные причины и механизмы
незавершенного фагоцитоза:
Уменьшение количества фагоцитов:
а) наследственные формы:
↓ гранулоцитопоэз → нейтропения;
гипоплазия селезенки → моноцитопения.
б) врожденная (аутоиммунная) нейтропения.
в) приобретенная нейтропения (при
заболеваниях со спленомегалией).
Неэффективный гранулоцитопоэз
(качественные изменения нейтрофилов):
а) нарушения подвижности фагоцитов
(нарушения обратимой полимеризации
сократительного белка фагоцитов —
актина). Пример:синдром «ленивых
лейкоцитов»
наследственная форма;
приобретенная форма (при ↑ сывороточного
IgE, ↓ энергии или ↓ цАМФ
в клетках).
б) нарушения образования фаголизосом
(наследственный дефект образования
лизосомальных гранул — их слияние →
гигантские гранулы). Пример: синдром
Чедиака–Хигаси
в) нарушение инактивации и разрушения
объектов фагоцитоза:
наследственный дефицит кислородзависимого
механизма бактерицидности (↓ НАДФ-оксидазы
и/или миелопероксидазы в фагоцитах).
Пример:хронический гранулематознаследственный дефицит кислороднезависимого
механизма бактерицидности (↓ лизоцима,
лактоферрина, катионных белков в
фагоцитах).
Нарушение распознавания объектов
фагоцитоза:
наследственный дефицит/дефект рецепторов
фагоцитаприобретенный дефицит опсонинов и
хемоаттрактантов (тяжелые болезни
печени, интоксикации и др.)
Нарушения нейрогормональной регуляции
фагоцитоза:
адреналин (в малых дозах) → ↑ фагоцитоз;
ацетилхолин → ↓ фагоцитоз;
глюкокортикоиды → ↓ фагоцитоз.
Синдром Чедиака-Хигаси
Тип наследования и частота.Наследуется
по аутосомно-рецессивному типу.
Генетический дефект и патогенез.
Мутация гена перемещения лизосом в
локус приводит к нарушениюLYST(lysosomaltraffilingregulator) и появлению гигантских
но функционально не активных гранул,
нарушается процесс поступления
миелопероксидазы в вакуоли, характерен
слабый ответ нейтрофилов на хемотакические
стимулы.
Клинические проявления.Гепатоспленомегалия, альбинизм
кожи, волос, глаз, фотофобия, лимфоаденопатия,
бронхиты, пневмонии, абсцессы в коже и
клетчатке, ретикулярные злокачественные
лимфомы
Диагностика. Гранулоцитопения,
нарушение хемотаксиса нейтрофилов,
фагоцитоза. Снижено количествоEK-клеток
и их функциональная активность.
Хроническая гранулематозная болезнь
Тип наследования и частота. Наиболее
часто встречается Х-сцепленная форма
(около 70%). Частота встречаемости около
1: 200 000.
Генетический дефект и патогенез.
Мутация одного из генов, ответственных
за синтез НАДФ∙Н. Вследствие этого
дефекта фагоциты пациентов не способны
продуцировать супероксидный радикал
и перекись водорода → отсутствие
кислородзависимых механизмов
бактерицидности. В результате
микроорганизмы внутри фагоцитов остаются
жизнеспособными, персистируют
внутриклеточно, вызывают иммунный ответ
с формированием гранулем. Гранулемы
могут нарушать функцию органов в которых
располагаются (задержка мочи, обструкция
кишечника).
Клинические проявления.Экзематозный дерматит, гнойные
поражения кожи, абсцессы в различных
органах, гепатоспленомегалия, лимфадениты,
бронхопневмонии, дерматиты, диарея,
грибковая инфекция.
Диагностика.Отсутствие или резкое
угнетение киллинга фагоцитированных
бактерий и грибов, отрицательные и
сниженные НСТ-тест (фагоциты больных
краситель не восстанавливают),
хемилюминесценция после фагоцитоза
частиц зимазана или латекса, хемотаксис
нейтрофилов.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Источник
СИНДРОМ “ЛЕНИВЫХ” ЛЕЙКОЦИТОВ МИЛЛЕРА
синдром ленивый лейкоцит
Недавно Миллер и Оски описали новый синдром, при котором глубокая периферическая нейтропения объясняется нарушенной подвижностью нейтрофилов, что препятствует их выходу из костного мозга и миграции к очагам инфекции.
Синдром «ленивых лейкоцитов» Миллера — совокупность наследственных дефектов функции нейтрофильных гранулоцитов:
- · Недостаточности миграционной активности (замедленный хемотаксис)
- · Снижения интенсивности фагоцитоза (замедленный хемотаксис и вялый фагоцитоз являются результатом нарушения функции цитоскелета, обеспечивающего локомоцию клетки и фагоцитоз)
- · Недостаточности бактерицидной функции, прежде всего из-за дефекта кислородного механизма.
У девочки 2 лет и у мальчика 5 лет абсолютное число гранулоцитов составляло соответственно 135 и 170 в 1 мм3 при нормальном состоянии костного мозга. Они страдали рецидивирующим стоматитом, гингивитом, отитом и вялотекущей лихорадкой с субфебрильной температурой. Слабая реакция на инъекцию бактериального пирогена указывала на нарушенный выброс из костного мозга “запасных сегментоядерных клеток”. Тесты in vitro показали слабую случайную и неслучайную (хемотактическую) подвижность нейтрофильных гранулоцитов. Фагоцитарная и бактерицидная активность была нормальной. Поэтому предполагается, что данная форма представляет собой не описанный ранее первичный дефект функции нейтрофилов, возможно, из-за дефекта мембраны сегментоядерных клеток.
В патогенезе «синдрома ленивых лейкоцитов» ведущими механизмами являются ограниченная подвижность гранулоцитов и их нарушенный выход из костного мозга.
К нарушению выхода зрелых лейкоцитов из костного мозга могут приводить несколько факторов. Во-первых, это может быть связано с генетически обусловленными дефектами сократительных белков, в частности с нарушением способности актина к полимеризации, что проявляется в нарушении двигательной активности нейтрофилов. Во-вторых, угнетение миграции нейтрофилов может быть следствием повреждения клеточных мембран наследственного или приобретенного характера, сопровождающегося снижением адгезивной способности, возможности лейкоцитов отвечать на хемотаксический стимул. И, наконец, причиной снижения подвижности нейтрофилов могут быть дефекты их энергетического обеспечения.
Предполагают, что в случае синдрома «ленивых лейкоцитов» есть дефект мембраны клетки, нормальное состояние которой является необходимым для каждого вида движения. Нарушение мембраны клетки нейтрофилов костного мозга у больных, имеющих синдром «ленивых лейкоцитов», удается проявить в замедленном их поступлении в воспалительные очаги (тест «кожного окна»).
Синдром «ленивых лейкоцитов» в сочетании с врождённой недостаточностью экзокринной функции поджелудочной железы называется болезнью Швамхмана (Швемкмана), с полным альбинизмом — болезнью Чедьяка–Хигаси (при этом заболевании в цитоплазме нейтрофилов, макрофагов, моноцитов и лимфоцитов обнаруживаются гигантские азурофильные гранулы, а в меланоцитах происходит патологическая агрегация меланосом, лежащая в основе альбинизма).
Источник
Синдром
«ленивых лейкоцитов» Миллера —
совокупность наследственных дефектов
функции нейтрофильных гранулоцитов:
Недостаточности
миграционной активности (замедленный
хемотаксис)
Снижения
интенсивности фагоцитоза (замедленный
хемотаксис и вялый фагоцитоз являются
результатом нарушения функции цитоскелета,
обеспечивающего локомоцию клетки и
фагоцитоз)
Недостаточности
бактерицидной функции, прежде всего
из-за дефекта кислородного механизма.
Синдром
«ленивых лейкоцитов» в сочетании с
врождённой недостаточностью экзокринной
функции поджелудочной железы называется
болезнью Шва́хмана (Шве́кмана), с полным
альбинизмом — болезнью Чедьяка—Хигаси
(при этом заболевании в цитоплазме
нейтрофилов, макрофагов, моноцитов и
лимфоцитов обнаруживаются гигантские
азурофильные гранулы, а в меланоцитах
происходит патологическая агрегация
меланосом, лежащая в основе альбинизма).
Дефекты фагоцитарных энзимов и цитоскелета
Кроме
того, описаны наследственные дефекты
ферментов фагоцитов и селективные
нарушения в функционировании элементов
цитоскелета:
Первичная
недостаточность миелопероксидазы и
других энзимовПервичный
дефект полимеризации актинаДефицит
тафцина.
Первичная
недостаточность миелопероксидазы
нейтрофильных гранулоцитов и
моноцитов/макрофагов наследуется
по аутосомно-рецессивному типу. При
этом не происходит синтеза из пероксида
водорода других активных метаболитов
кислорода (прежде всего гидроксильного
радикала) и галоид-содержащих соединений.
Эозинофильные гранулоциты не страдают.
Клинические проявления болезни
соответствуют хронической гранулематозной
болезни детей, но протекают значительно
менее тяжело, т.к. фагоциты не теряют
способность образовывать пероксид
водорода. Кроме того, известны
наследственные дефекты НАДН-оксидазы,
глютатион-пероксидазы,
глюкозо-6-фосфат-дегидрогеназы
нейтрофильных гранулоцитов.
Первичный
дефект полимеризации актина в нейтрофильных
гранулоцитах
характеризуется утратой ими способности
к локомоции и фагоцитозу вследствие
блокады процесса полимеризации актина,
необходимого для образования псевдоподий
и фагосом.
Наследственный
дефект образования тафцина
проявляется хроническими неспецифическими
воспалительными заболеваниями лёгких
и лимфоузлов. Синдром наследуется по
аутосомно-доминантному типу. Тафцин —
тетрапептид (тир—лиз—про—арг),
высвобождающийся из молекулы IgG под
воздействием специфических протеаз
фагоцитов; он усиливает фагоцитарную
активность нейтрофильных гранулоцитов.
Первичные
дефициты белков комплемента.
Недостаточность
белков комплемента проявляется по-разному
в зависимости от того, какой (или какие)
белки отсутствуют.
Выделяют
три группы заболеваний, связанных с
первичным дефицитом комплемента:
Комплемент-зависимые
иммунодефицитные синдромыКомплемент-ассоциированные
аутоиммунные болезниНаследственный
ангионевротический отёк Квинке—Ослера.
Комплемент-зависимые иммунодефицитные синдромы.
Комплемент-зависимые
иммунодефицитные синдромы — заболевания,
сопровождающиеся недостаточностью
антибактериальной защиты организма.
Они проявляются частыми инфекционными
процессами в различных органах и тканях.
Поскольку белки комплемента при активации
играют роль хемоаттрактантов и опсонинов,
обеспечивая эффективную функцию
фагоцитирующих клеток, то при дефиците
компонентов комплемента формируется
вторичная недостаточность функции
макрофагов и нейтрофильных гранулоцитов.
Особенно часто инфекционные процессы
при этом вызваны стрептококками, в
частности пневмококками, и Haemophilus
influenzae. В эту группу включают недостаточность
С3b-инактиватора, белков С3, С6 и С8.
Недостаточность
С3b-инактиватора. С3b-инактиватор играет
роль ингибитора альтернативного пути
активации комплемента. При его отсутствии
происходит быстрое потребление
С3-компонента (вторичный дефицит С3),
который в нормальных условиях принимает
активное участие в антибактериальной
защите организма. Белка С3 у больных в
плазме примерно 20% от нормы. Однако на
75% он представлен С3b-фрагментом. Уровень
нативного С3 составляет всего 5% от нормы.
Скорость расщепления С3 у больных
повышена почти в 5 раз. Показано, что
через 2 часа после инъекции нативного
С3 расщеплению подвергается 40% введённых
молекул. Помимо вторичного дефицита С3
формируется вторичная недостаточность
белка С5, однако она менее выражена
(примерно 40% от нормального уровня).
Заметно снижена концентрация фактора
В — 5% от нормы (расщепление фактора В
происходит под влиянием фактора D).
Уровень пропердина снижен незначительно.
Больные при этом заболевании страдают
различными бактериальными инфекциями.
Недостаточность
С3. Недостаточность
С3-компонента комплемента также
проявляется различными бактериозами.
В основе заболевания, в отличие от
недостаточности С3b-инактиватора, лежит
первичный дефицит С3-белка.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
01.06.201530.09 Mб23Епифанов Лечебная физкультура.pdf
- #
- #
- #
Источник
Ñîâîêóïíîñòü íàñëåäñòâåííûõ äåôåêòîâ ôóíêöèè íåéòðîôèëüíûõ ãðàíóëîöèòîâ. Ïàòîãåíåç ñèíäðîìà “ëåíèâûõ” ëåéêîöèòîâ Ìèëëåðà. Ôàêòîðû, ïðèâîäÿùèå ê íàðóøåíèþ âûõîäà çðåëûõ ëåéêîöèòîâ èç êîñòíîãî ìîçãà. Áîëåçíè, ñâÿçàííûå ñ ñèíäðîìîì “ëåíèâûõ” ëåéêîöèòîâ.
Ñòóäåíòû, àñïèðàíòû, ìîëîäûå ó÷åíûå, èñïîëüçóþùèå áàçó çíàíèé â ñâîåé ó÷åáå è ðàáîòå, áóäóò âàì î÷åíü áëàãîäàðíû.
Ðàçìåùåíî íà https://www.allbest.ru/
ÑÈÍÄÐÎÌ “ËÅÍÈÂÛÕ” ËÅÉÊÎÖÈÒÎÂ ÌÈËËÅÐÀ
ñèíäðîì ëåíèâûé ëåéêîöèò
Íåäàâíî Ìèëëåð è Îñêè îïèñàëè íîâûé ñèíäðîì, ïðè êîòîðîì ãëóáîêàÿ ïåðèôåðè÷åñêàÿ íåéòðîïåíèÿ îáúÿñíÿåòñÿ íàðóøåííîé ïîäâèæíîñòüþ íåéòðîôèëîâ, ÷òî ïðåïÿòñòâóåò èõ âûõîäó èç êîñòíîãî ìîçãà è ìèãðàöèè ê î÷àãàì èíôåêöèè.
Ñèíäðîì «ëåíèâûõ ëåéêîöèòîâ» Ìèëëåðà — ñîâîêóïíîñòü íàñëåäñòâåííûõ äåôåêòîâ ôóíêöèè íåéòðîôèëüíûõ ãðàíóëîöèòîâ:
· Íåäîñòàòî÷íîñòè ìèãðàöèîííîé àêòèâíîñòè (çàìåäëåííûé õåìîòàêñèñ)
· Ñíèæåíèÿ èíòåíñèâíîñòè ôàãîöèòîçà (çàìåäëåííûé õåìîòàêñèñ è âÿëûé ôàãîöèòîç ÿâëÿþòñÿ ðåçóëüòàòîì íàðóøåíèÿ ôóíêöèè öèòîñêåëåòà, îáåñïå÷èâàþùåãî ëîêîìîöèþ êëåòêè è ôàãîöèòîç)
· Íåäîñòàòî÷íîñòè áàêòåðèöèäíîé ôóíêöèè, ïðåæäå âñåãî èç-çà äåôåêòà êèñëîðîäíîãî ìåõàíèçìà.
Ó äåâî÷êè 2 ëåò è ó ìàëü÷èêà 5 ëåò àáñîëþòíîå ÷èñëî ãðàíóëîöèòîâ ñîñòàâëÿëî ñîîòâåòñòâåííî 135 è 170 â 1 ìì3 ïðè íîðìàëüíîì ñîñòîÿíèè êîñòíîãî ìîçãà. Îíè ñòðàäàëè ðåöèäèâèðóþùèì ñòîìàòèòîì, ãèíãèâèòîì, îòèòîì è âÿëîòåêóùåé ëèõîðàäêîé ñ ñóáôåáðèëüíîé òåìïåðàòóðîé. Ñëàáàÿ ðåàêöèÿ íà èíúåêöèþ áàêòåðèàëüíîãî ïèðîãåíà óêàçûâàëà íà íàðóøåííûé âûáðîñ èç êîñòíîãî ìîçãà “çàïàñíûõ ñåãìåíòîÿäåðíûõ êëåòîê”. Òåñòû in vitro ïîêàçàëè ñëàáóþ ñëó÷àéíóþ è íåñëó÷àéíóþ (õåìîòàêòè÷åñêóþ) ïîäâèæíîñòü íåéòðîôèëüíûõ ãðàíóëîöèòîâ. Ôàãîöèòàðíàÿ è áàêòåðèöèäíàÿ àêòèâíîñòü áûëà íîðìàëüíîé. Ïîýòîìó ïðåäïîëàãàåòñÿ, ÷òî äàííàÿ ôîðìà ïðåäñòàâëÿåò ñîáîé íå îïèñàííûé ðàíåå ïåðâè÷íûé äåôåêò ôóíêöèè íåéòðîôèëîâ, âîçìîæíî, èç-çà äåôåêòà ìåìáðàíû ñåãìåíòîÿäåðíûõ êëåòîê.
 ïàòîãåíåçå «ñèíäðîìà ëåíèâûõ ëåéêîöèòîâ» âåäóùèìè ìåõàíèçìàìè ÿâëÿþòñÿ îãðàíè÷åííàÿ ïîäâèæíîñòü ãðàíóëîöèòîâ è èõ íàðóøåííûé âûõîä èç êîñòíîãî ìîçãà.
Ê íàðóøåíèþ âûõîäà çðåëûõ ëåéêîöèòîâ èç êîñòíîãî ìîçãà ìîãóò ïðèâîäèòü íåñêîëüêî ôàêòîðîâ. Âî-ïåðâûõ, ýòî ìîæåò áûòü ñâÿçàíî ñ ãåíåòè÷åñêè îáóñëîâëåííûìè äåôåêòàìè ñîêðàòèòåëüíûõ áåëêîâ, â ÷àñòíîñòè ñ íàðóøåíèåì ñïîñîáíîñòè àêòèíà ê ïîëèìåðèçàöèè, ÷òî ïðîÿâëÿåòñÿ â íàðóøåíèè äâèãàòåëüíîé àêòèâíîñòè íåéòðîôèëîâ. Âî-âòîðûõ, óãíåòåíèå ìèãðàöèè íåéòðîôèëîâ ìîæåò áûòü ñëåäñòâèåì ïîâðåæäåíèÿ êëåòî÷íûõ ìåìáðàí íàñëåäñòâåííîãî èëè ïðèîáðåòåííîãî õàðàêòåðà, ñîïðîâîæäàþùåãîñÿ ñíèæåíèåì àäãåçèâíîé ñïîñîáíîñòè, âîçìîæíîñòè ëåéêîöèòîâ îòâå÷àòü íà õåìîòàêñè÷åñêèé ñòèìóë. È, íàêîíåö, ïðè÷èíîé ñíèæåíèÿ ïîäâèæíîñòè íåéòðîôèëîâ ìîãóò áûòü äåôåêòû èõ ýíåðãåòè÷åñêîãî îáåñïå÷åíèÿ.
Ïðåäïîëàãàþò, ÷òî â ñëó÷àå ñèíäðîìà «ëåíèâûõ ëåéêîöèòîâ» åñòü äåôåêò ìåìáðàíû êëåòêè, íîðìàëüíîå ñîñòîÿíèå êîòîðîé ÿâëÿåòñÿ íåîáõîäèìûì äëÿ êàæäîãî âèäà äâèæåíèÿ. Íàðóøåíèå ìåìáðàíû êëåòêè íåéòðîôèëîâ êîñòíîãî ìîçãà ó áîëüíûõ, èìåþùèõ ñèíäðîì «ëåíèâûõ ëåéêîöèòîâ», óäàåòñÿ ïðîÿâèòü â çàìåäëåííîì èõ ïîñòóïëåíèè â âîñïàëèòåëüíûå î÷àãè (òåñò «êîæíîãî îêíà»).
Ñèíäðîì «ëåíèâûõ ëåéêîöèòîâ» â ñî÷åòàíèè ñ âðîæä¸ííîé íåäîñòàòî÷íîñòüþ ýêçîêðèííîé ôóíêöèè ïîäæåëóäî÷íîé æåëåçû íàçûâàåòñÿ áîëåçíüþ Øâàìõìàíà(Øâåìêìàíà), ñ ïîëíûì àëüáèíèçìîì — áîëåçíüþ ×åäüÿêà–Õèãàñè (ïðè ýòîì çàáîëåâàíèè â öèòîïëàçìå íåéòðîôèëîâ, ìàêðîôàãîâ, ìîíîöèòîâ è ëèìôîöèòîâ îáíàðóæèâàþòñÿ ãèãàíòñêèå àçóðîôèëüíûå ãðàíóëû, à â ìåëàíîöèòàõ ïðîèñõîäèò ïàòîëîãè÷åñêàÿ àãðåãàöèÿ ìåëàíîñîì, ëåæàùàÿ â îñíîâå àëüáèíèçìà).
Ðàçìåùåíî íà Allbest.ru
…
Источник
Иммунодефицит возникает, когда один или несколько компонентов иммунной системы являются дефектными. Наиболее распространенной причиной иммунодефицита является недоедание. Однако в развитых странах большинство болезней иммунодефицита унаследованы, и их обычно наблюдают как рецидивирующие или распространенные инфекции у очень маленьких детей. Менее часто приобретенные иммунные недостатки с причинами, отличными от недоедания, могут возникать позже. Хотя патогенез многих из этих приобретенных нарушений остается неясным, некоторые из них вызваны известными агентами, такими как лекарственные средства или радиация, которые повреждают лимфоциты, корь или ВИЧ-инфекция. Изучая, какие инфекции сопровождают определенный унаследованный или приобретенный иммунный дефицит, можно определить, какие компоненты иммунной системы важны в ответ на некоторые инфекционные агенты. Унаследованные заболевания иммунодефицита также показывают, как взаимодействие между различными типами клеток способствует иммунному ответу и развитию Т- и В-лимфоцитов.
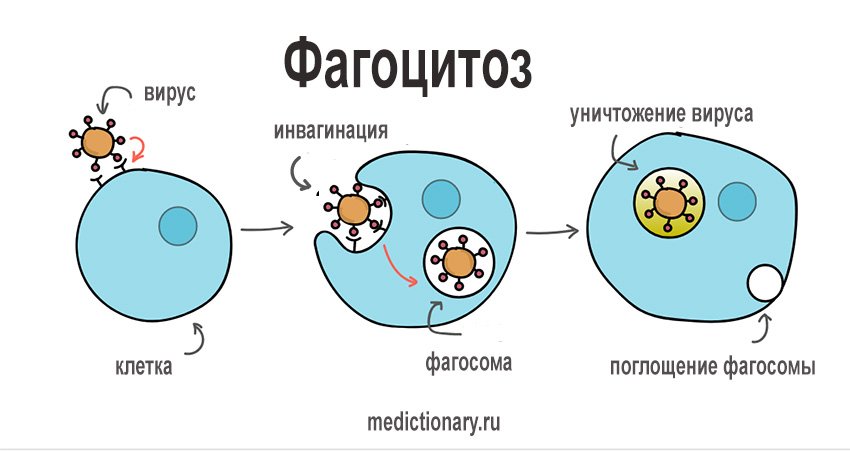
Иммунодефицитные синдромы, связанные с дефектом фагоцитоза, обычно предрасполагают пациентов к инфекциям. Чувствительность к инфекции фагоцитарной дисфункции варьируется от мягких рецидивирующих кожных инфекций до преимущественно фатальной системной инфекции. Затронутые пациенты более восприимчивы к бактериальным и грибковым инфекциям, но имеют нормальную устойчивость к вирусным инфекциям. Большинство синдромов иммунодефицита, связанных с дефектом фагоцитоза, диагностируются в младенчестве из-за тяжести инфекции.
Иммунодефицитные синдромы, связанные с дефектом фагоцитоза, можно разделить на внешние и внутренние дефекты:
- Наружные дефекты включают аномалии, вторичные по отношению к недостаткам антител и факторам комплемента. Внешние факторы могут приводить к нейтропении путем подавления образования гранулоцитов или снижения количества циркулирующих нейтрофилов с помощью аутоантител лейкоцитов или изотиобластов, направленных против нейтрофильных антигенов.
- Внутренние нарушения гранулоцитов можно разделить на те, которые возникают в результате дефектов в способности убивать гранулоциты и те, которые ингибируют хемотаксис (движение клеток). Внутренние нарушения фагоцитарной способности включают хроническое гранулематозное заболевание, синдром Чедиак-Хигаши и специфический дефицит гранулезы.
В разделе синдромов иммунодефицита, связанных с дефектом фагоцитоза, будет рассмотрено более подробное хроническое гранулематозное заболевание.
Хроническая гранулематозная болезнь является первичной иммунной недостаточностью, которая поражает фагоциты врожденной иммунной системы и приводит к рецидивирующим или стойким внутриклеточным бактериальным и грибковым инфекциям и к образованию гранулем. Приблизительно две трети пациентов испытывают первые симптомы в первый год жизни в виде инфекций, дерматитов (иногда при рождении), желудочно-кишечных осложнений (обструкция или прерывистая кровавая диарея из-за колита).
Фагоциты (нейтрофилы и макрофаги) требуют, чтобы фермент продуцировал реактивные виды кислорода, чтобы уничтожить бактерии после глотания (фагоцитоз), процесс, известный как респираторный взрыв. Этот фермент называется фагоцитарной никотинамидадениндинуклеотидфосфатоксидазой. Этот фермент окисляет никотинамидадениндинуклеотидфосфат и уменьшает молекулярный кислород для получения супероксидных анионов — реактивного кислорода. Затем супероксид диспропорционирован в пероксиде и молекулярном кислороде супероксиддисмутазой. Наконец, пероксид используется миелопероксидазой для окисления ионов хлорида до гипохлорита, который токсичен для бактерий. Таким образом, никотинамидадениндинуклеотид-фосфат-оксидаза имеет решающее значение для фагоцитарного уничтожения бактерий реактивными кислородными видами.
Хроническая гранулематозная болезнь представляет собой генетическое гетерогенное нарушение иммунодефицита из-за неспособности фагоцитов убивать зародыши, которые они проглотили. Это повреждение вызвано некоторыми дефектами комплекса никотинамидадениндинуклеотидфосфатоксидазы, которая вызывает микробицидное нарушение дыхания. При хроническом гранулематозном заболевании фагоциты обычно проглатывают бактерии, но они не могут их убить.
Пациенты с этим заболеванием восприимчивы к тяжелым и рецидивирующим инфекциям из-за каталаза-положительных организмов и организмов, устойчивых к неоадъювантным убийствам. Каталазо-отрицательные бактерии, такие как стрептококки и пневмококки, которые обладают способностью генерировать перекись водорода, погибают, как обычно. Внутриклеточная выживаемость проглоченных бактерий приводит к развитию гранулемы в лимфатических узлах, коже, легких, печени, желудочно-кишечном тракте и / или костях.
Хроническое гранулематозное заболевание обычно унаследовано в Х-связанном рецессивном способе. Большинство пациентов (приблизительно 80%) являются мужчинами, у которых есть химерные мутации на X-связанном гене. Ген, ответственный за эту форму заболевания, описан в области р21.1 Х-хромосомы. Однако в подтипах хронических гранулематозных заболеваний аутосомно-рецессивные формы могут быть связаны с менее тяжелым заболеванием. Степень, в которой экологические и вторичные генетические факторы влияют на фенотипическую экспрессию заболевания, неизвестна.
Хроническое гранулематозное заболевание характеризуется гистологически смешанным гнойным и гранулематозным воспалением. Типичной особенностью висцеральных гранулем является наличие золотисто-коричневых пигментных гистиоцитов. Гистохимические пятна показывают, что этот материал состоит из ненасыщенных жирных кислот, фосфолипидов и гликопротеинов.
Периодическое окрашивание реагентом Шиффа (реакция PAS) указывает на присутствие углеводов, в частности полисахаридов, таких как мукопротеины. Эти вещества окрашиваются красновато-фиолетовыми с реакцией PAS.
Электронные микроскопические данные свидетельствуют о том, что пигмент представляет собой тело липофусцина и, по-видимому, производится из лизосом. Гранулемы состоят из нейтрофилов и макрофагов, которые содержат желтые включения с участками некроза.
Источник