
Митохондриальная энцефалопатия код мкб

MELAS-синдром – это генетическое заболевание с поражением центральной нервной системы, мышечной ткани и различных органов. В основе патологии лежит нарушение тканевого дыхания и дефект энергетического метаболизма. Клиническая картина отличается гетерогенностью, включает острые эпизоды, напоминающие инсульт, эпилептические приступы, непереносимость физических нагрузок вследствие мышечной слабости. Для диагностики применяются МРТ, ЭЭГ, ЭНМГ и другие исследования. Окончательный диагноз устанавливается при обнаружении точечных мутаций в митохондриальной ДНК. Лечение заключается в назначении метаболических препаратов и симптоматической терапии.
Общие сведения
MELAS-синдром (митохондриальная энцефалопатия с лактат-ацидозом и инсультоподобными эпизодами) относится к болезням, обусловленным генетическим дефектом митохондриальной ДНК. При этом синдроме нарушается энергопродукция в митохондриальной дыхательной цепи. Заболевание впервые было описано в 1984 году. По разным данным, патология встречается с частотой от 1:15 000 до 1:20 000 человек. Гендерные различия отсутствуют. Средний возраст дебюта – 6-10 лет.
MELAS-синдром
Причины
Причиной возникновения патологии являются точечные мутации митохондриальной ДНК. На сегодняшний день известно около 10 генов, при дефекте которых наблюдается манифестация MELAS-синдрома. Наиболее часто выявляются мутации в генах, кодирующих транспортную РНК. Чаще всего (80-90% случаев) обнаруживается мутация A3243G в гене MTTL1 транспортной РНК аминокислоты лейцина.
Из-за дефекта нарушается синтез митохондриальных белков. Наличие мутации необязательно гарантирует фенотипическое проявление заболевания, что связано с феноменом гетероплазмии, то есть, одновременного наличия в клетке, органе или организме нормальной и мутантной митохондриальных ДНК. Большое количество дефектных ДНК повышает вероятность клинической манифестации синдрома.
Патогенез
Генетически детерминированный дефект синтеза митохондриальных белков (ферментов тканевого дыхания) приводит к нарушению процессов окислительного фосфорилирования – последней стадии расщепления биологических субстратов: жирных кислот, углеводов. Окислительное фосфорилирование считается главным звеном энергетического метаболизма, обеспечивает образование подавляющего количества энергоносителей – молекул АТФ.
Сбой этих процессов происходит в каждой митохондрии с дефектной ДНК. Поскольку митохондрии представлены практически во всех клетках организма, поражение носит мультисистемный характер. Сильнее всего страдают органы и ткани с высокой энергетической потребностью: центральная и периферическая нервная система, миокард, скелетные мышцы.
В мышцах энергетический дефицит (тканевая гипоксия) приводит к избыточному образованию молочной кислоты. В генезе острых церебральных нарушений лежит дефицит оксида азота вследствие накопления в гладких мышцах сосудов фермента цитохромоксидазы. При недостаточности оксида азота возникает вазоконстрикция, агрегация тромбоцитов.
Классификация
По выраженности клинических проявлений выделяют 3 формы MELAS-синдрома:
- Бессимптомное носительство – наличие генетической мутации и изменений при биопсии мышц на фоне полного отсутствия клинических признаков заболевания.
- Олигосимптоматическая – обнаруживаются отдельные компоненты синдрома: мышечная слабость, головные боли и пр.
- Манифестная – яркая клиническая картина с острыми эпизодами.
Симптомы
Обычно вначале манифестируют неврологические симптомы, которые возникают уже в 6-15 лет. Наиболее типичными считаются инсультоподобные эпизоды (метаболический инсульт). Частые проявления – гемианопсия (выпадение половины поля зрения), нарушение равновесия, восприятия или воспроизведения речи, изменения сознания.
У половины пациентов отмечаются фокальные или генерализованные тонико-клонические эпилептические припадки, головные боли, напоминающие мигрень (односторонние, пульсирующие). Характерные черты таких приступов – возраст больных менее 40 лет, провоцирование инфекционным заболеванием, быстрый регресс симптоматики и частое рецидивирование.
Реже наблюдаются острые психозы и гемипарезы. Из других неврологических проявлений можно выделить задержку нервно-психического развития, заторможенность, когнитивные нарушения. Вследствие атрофии зрительных нервов постепенно ухудшается зрение. Еще один специфичный признак MELAS-синдрома – выраженная мышечная слабость и плохая переносимость физической нагрузки.
Часто встречаются боли, судороги в мышцах. Мышечные спазмы также могут вызываться снижением концентрации кальция в крови вследствие гипопаратиреоза. У многих пациентов наблюдается постепенное ухудшение слуха (нейросенсорная тугоухость), возникает сахарный диабет. Кардиологические признаки заболевания включают кардиомиопатии, аритмии, хроническую сердечную недостаточность.
Осложнения
MELAS-синдром является тяжелым заболеванием, характеризующимся широким спектром неблагоприятных последствий. Наиболее частые осложнения, приводящие к летальному исходу, связаны с митохондриальной энцефалопатией. К ним относятся повторные «метаболические инсульты» и эпилептические статусы. Небольшая часть пациентов впадает в кому.
У больных повышен риск развития жизнеугрожающих нарушений ритма (желудочковая тахикардия, фибрилляция), остановки сердца. Нейросенсорная тугоухость и атрофия зрительного нерва могут привести к полной потере слуха и зрения. В единичных случаях возникает кишечная непроходимость, хроническая почечная и печеночная недостаточность.
Диагностика
Больных с MELAS-синдромом курируют врачи-неврологи, педиатры. При общем осмотре обращает на себя внимание низкий рост, выраженная гипотония, гипотрофия мышц. Заподозрить заболевание позволяет сочетание миопатии с острыми неврологическими расстройствами у пациента молодого возраста. Дополнительное обследование, направленное на уточнение диагноза, включает:
- Лабораторные исследования. В биохимическом анализе крови определяется высокий уровень молочной кислоты, глюкозы и гликированного гемоглобина, уменьшение концентрации кальция. При анализе крови на гормоны обнаруживают снижение содержания соматотропного и паратиреоидного гормонов. В общем анализе мочи часто выявляется протеинурия.
- МРТ головного мозга. Повреждения мозга визуализируются как очаги неправильной формы с четкими контурами и слабо повышенной интенсивностью МР-сигнала. Отмечаются признаки объемного воздействия на субарахноидальное пространство. Нередко выявляются кальцинаты в базальных ганглиях с преимущественной локализацией в затылочной и теменной областях. При повторном проведении МРТ обнаруживается флюктуация или исчезновение очагов.
- МР-спектроскопия. Магнитно-резонансная спектроскопия головного мозга показывает значительное снижение высокоэнергетических фосфорных соединений и увеличение концентрации лактата.
- ЭЭГ. На электроэнцефалограмме просматривается замедление основной активности, диффузные нарушения биоэлектрических процессов головного мозга, признаки островолновой активности, усиливающейся на фоне гипервентиляции.
- ЭНМГ. Электронейромиография подтверждает наличие неспецифических признаков – сокращение длительности потенциала и амплитуды двигательных единиц, блок и замедление проведения нервного импульса.
- Биопсия мышц. Наиболее типичные морфологические изменения – феномен «рваных красных волокон» (миофибриллы с большим числом пролиферирующих измененных митохондрий), склероз перимизия, регионарные некрозы. Отмечается положительная гистохимическая окраска на цитохромоксидазу и сукцинатдегидрогеназу.
- Исследование ДНК. Решающий диагностический тест для верификации синдрома MELAS. В лимфоцитах периферической крови чаще других выявляется мутация A3243G гена MTTL1.
Дифференциальный диагноз острых эпизодов проводят с ишемическими инсультами, субарахноидальным кровоизлиянием, острыми инфекционными менингоэнцефалитами. Миопатический синдром следует отличать от наследственных мышечных дистрофий, полимиозита. Патологию также нужно дифференцировать с другими митохондриальными заболеваниями.
Лечение MELAS-синдрома
Радикальное лечение не разработано. Все методы терапии направлены на улучшение состояния пациента и носят лишь паллиативный характер. Необходима диета с ограничением содержания углеводов. Диетическое питание требуется для снижения уровня глюкозы, негативно влияющей на параметры энергетического обмена, и для коррекции сахарного диабета. Применяются следующие медикаменты:
- Метаболические препараты. Для улучшения окислительного фосфорилирования в митохондриях назначается комплексное энерготропное лечение, включающее лекарства, способствующие переносу электронов в дыхательной цепи (коэнзим Q10, янтарная кислота), кофакторы ферментов энергетического обмена (рибофлавин, никотинамид), антиоксиданты (аскорбиновая кислота, токоферол).
- Предшественники и донаторы оксида азота. Во время инсультоподобного эпизода эффективными оказались медикаменты, повышающие уровень оксида азота в крови (L-аргинин, цитруллин), способствующие вазодилатации и улучшению микроциркуляции.
- Препараты для коррекции КЩР. Средства, снижающие уровень лактата в крови (корректоры лактат-ацидоза) используются только в остром периоде у пациентов с очень высоким содержанием в крови молочной кислоты. Их назначение требует большой осторожности из-за способности оказывать токсическое действие на нервную ткань. Возможно внутривенное введение гидрокарбоната натрия.
- Глюкокортикостероиды. Применение гормонов коры надпочечников (преднизолон) приводит к значительному регрессу неврологической симптоматики.
- Средства для коррекции гормональных нарушений. При развитии у сахарного диабета рекомендованы сахароснижающие препараты (метформин, глибенкламид), а при их неэффективности – инсулинотерапия. При возникновении гипопаратиреоза в схему лечения добавляют кальций и витамин Д.
- Противоэпилептические препараты. Широко используемые в эпилептологии производные вальпроевой кислоты строго противопоказаны, поскольку ингибируют энергетический метаболизм. Предпочтение отдается клоназепаму, ламотриджину, топирамату.
В зависимости от клинической картины назначают кардиотропные средства (бета-блокаторы, антиаритмики, ингибиторы АПФ), корректоры печеночной недостаточности (альбумин и медикаменты, подавляющие образование аммиака), сеансы гемодиализа. Следует максимально избегать применения препаратов, угнетающих митохондриальную функцию (барбитураты, хлорамфеникол, статины), поскольку это приводит к ухудшению состояния больного.
Прогноз и профилактика
Синдром MELAS – тяжелое прогрессирующее заболевание с высокой частотой летальных исходов. Продолжительность жизни у людей с бессимптомным носительством и олигосимптоматической формой не отличается от таковой в общей популяции. При манифестной форме, по различным данным, срок жизни составляет от 7 до 40 лет с момента дебюта болезни.
Методы первичной профилактики не разработаны. Поскольку неврологические приступы достаточно часто провоцируются острой инфекцией, для их предотвращения рекомендуется постановка прививок от гриппа и меры общей профилактики, направленные на повышение сопротивляемости организма (регулярные физические нагрузки, закаливание).
Источник
Связанные заболевания и их лечение
Описания заболеваний
Стандарты мед. помощи
Содержание
- Описание
- Причины
- Симптомы
- Лечение
- Основные медицинские услуги
- Клиники для лечения
Названия
Митохондриальные заболевания.
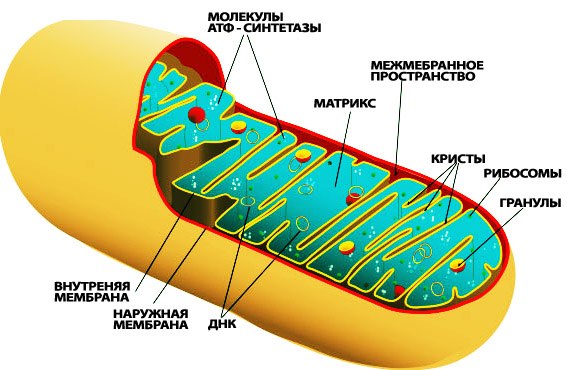
Строение митохондрии
Описание
Митохондриальные заболевания — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках эукариот, в частности, человека.
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами митохондрий, приводящими к нарушениям тканевого дыхания. Они передаются только по женской линии к детям обоих полов, так как сперматозоиды передают зиготе половину ядерного генома, а яйцеклетка поставляет и вторую половину генома, и митохондрии. Патологические нарушения клеточного энергетического обмена могут проявляться в виде дефектов различных звеньев в цикле Кребса, в дыхательной цепи, процессах бета-окисления.
Не все ферменты и другие регуляторы, необходимые для эффективного функционирования митохондрий, кодируются митохондриальной ДНК. Большая часть митохондриальных функций контролируется ядерной.
Можно выделить две группы митохондриальных заболеваний:
Ярко выраженные наследственные синдромы, обусловленные мутациями генов, ответственных за митохондриальные белки (синдром Барта, синдром Кернса-Сейра, синдром Пирсона, синдром MELAS, синдром MERRF и другие).
Вторичные митохондриальные заболевания, включающие нарушение клеточного энергообмена как важное звено формирования патогенеза (болезни соединительной ткани, синдром хронической усталости, гликогеноз, кардиомиопатия, мигрень, печёночная недостаточность, панцитопения, а также гипопаратиреоз, диабет, рахит и другие).
Причины
Повреждение митохондрий в основном возникаетиз-за воздействия реактивных форм кислорода (РФК). В настоящее время считают, что большинство РФК образуется комплексами I и III, вероятно, вследствие высвобождения электронов под воздействием НАД-Н и ФАД-Н в ЦПЭ. Митохондрии используют приблизительно 85% кислорода, потребляемого клеткой, в процессе образования АТФ. В ходе нормального процессаОФ от 0,4% до 4,0% всего употребляемого кислорода преобразуется в митохондриях в супероксидные радикалы (О2-). Супероксид трансформируется до пероксида водорода (Н2О2) с помощью ферментов детоксикации-марганцевой супероксиддисмутазы (Mn-СОД) или цинк/медь- супероксиддисмутазы (Cu/Zn СОД),- а затем до воды с помощью глутатионпероксидазы (ГП) или пероксидредоксина III (ПР III). Однако, если эти ферменты не способны достаточно быстроконвертировать РФК, такие как супероксид-радикал, до воды, происходит оксидативное повреждение и аккумулируется в митохондриях. Глутатион в ПР является одним из основных антиоксидантов в организме. Глутатион представляет собой трипептид, содержащий глутамин, глицин и цистеин. ГП требует селен в качестве кофактора.
Показано, сто супероксид in vitro повреждает железо-серный кластер, находящийся в в активном центре аконитазы, фертента цикла ТКК. Из-за этого железо вступает в реакцию с Н2О2 с образованием гидроксильных радикалов через реакцию Фентона (Fenton). Кроме того, оксид азота (NO) образуется в митохондриях с помощью митохондриальной синтазы оксида азота (МтСОА), а также свободно диффундирует в митохондрии из цитозоля. NO реагирует с O2 с образованием другого радикала- пероксинитрита (ONOO-). Вместе эти два радикала и другие радикалы могут нанести существенное повреждение митохондриям и другим компонентам клетки.
В митохондриях элементами, которые особенно подвержены воздействию свободных радикалов, являются липиды, белки, окислительно-восстановительные ферменты и мтДНК. Прямое повреждение митохондриальных белков снижает их аффинность к субстратам или коферментам и таким образом нарушают их функцию. Проблема осложняется тем, что если повреждение митохондрии произошло, то функция митохондрии может быть скомпрометирована увеличением потребностей клетки для процессов репарации энергии. Митохондриальная дисфункция может привести к цепному процессу, при котором митохондриальное повреждение влечет за собой дополнительное повреждение.
Комплекс I особенно чувствителен к воздействию оксида азота (NO). У животных, которым вводили природные и синтетические антагонисты комплекса I, как правило, наблюдается гибель нейронов. Нарушение функции комплекса I было ассоциировано с наследственной оптической нейропатией Лебера, болезнью Паркинсона и другими нейродегенеративными состояниями.
Гипергликемия индуцирует образование супероксида в митохондриях эндотелиальными клетками, который является важным медиатором диабетических осложнений, таких как сердечно- сосудистые заболевания. Образование супероксида в эндотелии также способствует развитию атеросклероза, гипертензии, сердечной недостаточности, старения, сепсиса, ишемически- реперфузионных повреждений и гиперхолестеринемии.
Медиаторы воспаления, такие как фактор некроза опухолей α (ФНОα) in vitro были связаны с митохондриальной дисфункциейи повышали образование ФРК. В модели застойной сердечной недостаточностидобавление ФНОα к культуре кардиомиоцитов повышало образование РФК и гипертрофию миоцитов. ФНОα вызывает митохондриальную дисфункциюпутем восстановления активности комплекса III в ЦПЭ, увеличивая образование РФК и повреждение мтДНК.
Дефицит питательных веществ или их избыток также может привести к митохондриальной дисфункции. Витамины, минералы и другие метаболиты работают как необходимые кофакторы для синтеза и функционирования митохондриальных ферментов и других составляющих, которые поддерживают функцию митохондрий, и диета с недостатком микрокомпонентов можетускорять старение митохондрий и способствовать нейродегенерации. Например, ферменты участвующие в цепи синтеза гемма, требуют достаточных количеств пиридоксина, железа, меди, цинка и рибофлавина. Недостаток питательных веществ, необходимых для каких- либо компонентов цикла ТКК или ЦПЭ, может привести к увеличению образования свободных радикалов и повреждению мтДНК.
Хорошо известно, что недостаток питательных веществ является широко распространенной причиной патогенеза многих заболеваний и является главным предметом спора в здравоохранении. Недостаток железа главным посредником в развитии общего груза заболеваний, затрагивающих приблизительно 2 миллиарда людей, преимущественно женщин и детей. Это наиболее распространенный тип дефицита питательных веществ. Низкий статус содержания железа снижает активность митохондрийпутем выключения комплекса IV и увеличенияоксидативного стресса. Механизмы, лежащие в основе процесса влияния дефицита питательных веществ (и в некоторых случаях избыток, как при перегрузке железом) на возникновение, развитие и прогрессирование заболеваний, возникающих вследствие нарушения митохондриальных функций, к настоящему времени уже изучены.
Наследование митохондриальных болезней:
Митохондрии наследуются иначе, чем ядерные гены. Ядерные гены в каждой соматической клетке обычно представлены двумя аллелями (за исключением большинства сцепленных с полом генов у гетерогаметного пола). Один аллель унаследован от отца, другой от матери. Однако митохондрии содержат собственную ДНК, причем в каждой митохондрии человека обычно содержится от 5 до 10 копий кольцевой молекулы ДНК ( Гетероплазмия), и все митохондрии наследуются от матери. Когда митохондрия делится, копии ДНК случайным образом распределяются между ее потомками. Если только одна из исходных молекул ДНК содержит мутацию, в результате случайного распределения такие мутантные молекулы могут накопиться в некоторых митохондриях. Митохондриальная болезнь начинает проявляться в тот момент, когда заметное число митохондрий во многих клетках данной ткани приобретают мутантные копии ДНК (пороговая экспрессия).
Мутации в митохондриальной ДНК происходят, по разным причинам, намного чаще, чем в ядерной. Это означает, что митохондриальные болезни достаточно часто проявляются из-за спонтанных вновь возникающих мутаций. Иногда темп мутирования увеличивается из-за мутаций в ядерных генах, кодирующих ферменты, которые контролируют репликацию ДНК митохондрий.
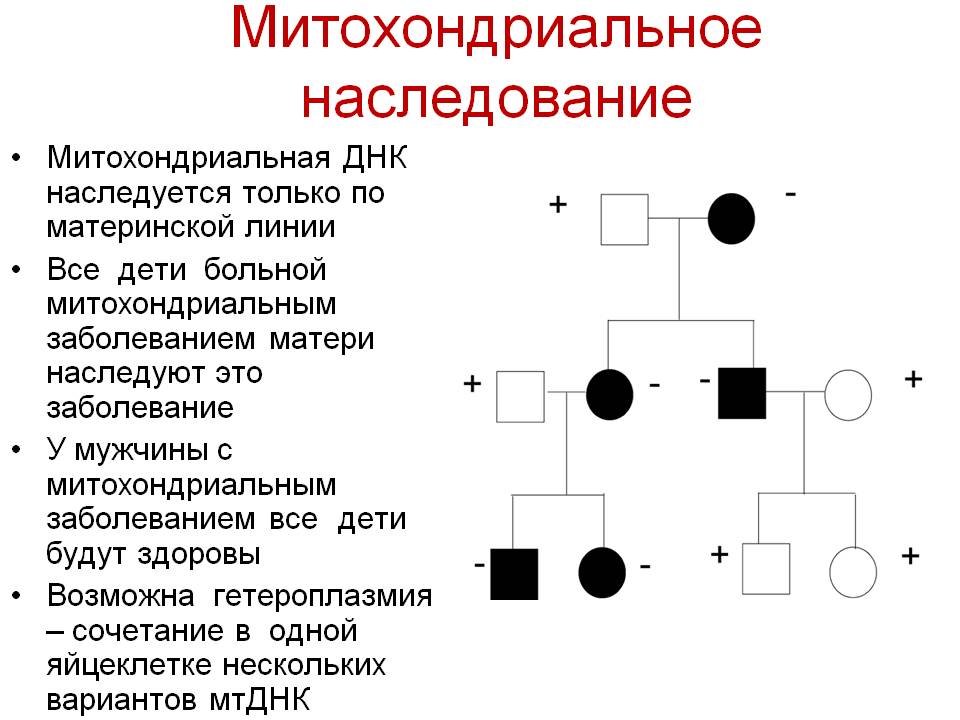
Генетические аспекты митохондриальных болезней
Симптомы
Эффекты митохондриальных заболеваний очень разнообразны. Из-за различного распределения дефектных митохондрий в разных органах мутация у одного человека может привести к заболеванию печени, а у другого — к заболеванию мозга. Величина проявления дефекта может быть большой или малой, и она может существенно изменяться, медленно нарастая во времени. Некоторые небольшие дефекты приводят лишь к неспособности пациента выдерживать физическую нагрузку, соответствующую его возрасту, и не сопровождаются серьёзными болезненными проявлениями. Другие дефекты могут быть более опасны, приводя к серьёзной патологии.
В общем случае митохондриальные заболевания проявляются сильнее при локализации дефектных митохондрий в мышцах, мозге, нервной ткани, поскольку эти органы требуют больше всего энергии для выполнения соответствующих функций.
Несмотря на то, что протекание митохондриальных заболеваний сильно отличаются у разных пациентов, на основании общих симптомов и конкретных мутаций, вызывающих болезнь, выделено несколько основных классов этих заболеваний.
Помимо относительно распространённой митохондриальной миопатии, встречаются:
1. Митохондриальный сахарный диабет, сопровождающийся глухотой (DAD, MIDD, синдром MELAS) — это сочетание, проявляющееся в раннем возрасте, может быть вызвано мутацией митохондриального гена MT-TL1, но сахарный диабет и глухота могут быть вызваны как митохондриальными заболеваниями, так и иными причинами;
2. Наследственная оптическая нейропатия Лебера, характеризующийся потерей зрения в раннем пубертатном периоде;
3. Синдром Вольфа-Паркинсона-Уайта;
4. Рассеянный склероз и подобные ему заболевания;
5. Синдром Лея (Leigh) или подострая некротизирующая энцефаломиопатия : после начального нормального постнатального развития болезнь проявляется обычно в конце первого года жизни, иногда — во взрослом возрасте. Болезнь сопровождается быстрой потерей функций организма и характеризуется судорогами, нарушенным состоянием сознания, деменцией, остановкой дыхания;
6. Нейропатия, атаксия, retinitis pigmentos и птоз: прогрессирующие симптомы нейропатии, атаксии, туннельное зрение и потеря зрения, птоз, деменция;
7. Митохондриальная нейрогастроинтенстинальная энцефалопатия: гастроинтестинальная псевдообструкция и кахексией, нейропатия, энцефалопатия с изменениями белого вещества головного мозга.
Лечение
В настоящее время лечение митохондриальных заболеваний находится в стадии разработки, но распространённым терапевтическим методом служит симптоматическая профилактика с помощью витаминов. В частности, в лечении синдрома MELAS у ряда пациентов оказались эффективными кофермент Q, который применяется как цитопротектор и антиоксидант при кардиомиопатиях и хронической сердечной недостаточности, рибофлавин и никотинамид. Также в качестве одного из методов применяются пируваты.
В настоящее время проводятся экспериментальные работы по изучению возможности экстракорпорального (in vitro) оплодотворения с использованием химерной яйцеклетки, ядро которой получено из яйцеклетки пациентки с митохондриальным заболеванием, а цитоплазму из другой яйцеклетки от женщины с нормально функционирующими митохондриями (замена ядра).
Основные медуслуги по стандартам лечения | ||
Клиники для лечения с лучшими ценами
|
Источник