
Митохондриальный синдром у ребенка что это такое

Генетическая информация очень нестабильна. Один из основных постулатов генетики гласит, что изменчивость — это основной фактор развития всего живого. Мутации необходимы для выживания вида. Однако некоторые изменчивости, в частности в митохондриях, приводят к негативным видоизменениям в генетической природе. Такова причина заболевания, носящее название митохондриальный синдром.
Такие заболевания не так часто встречаются, но исход большинства синдромов митохондриальной ДНК крайне неблагоприятен.
Митохондрии. Их функции в клетке
Напомним биологические азы. Митохондрия — это органелла в человеческой клетке, у которой наличествует свой ДНК-код. Передается всегда митохондрия от матери. Несет ее в себе материнская яйцеклетка. Митохондрии самостоятельно делятся в клетке, и многократно повторяют свой набор ДНК, копий которого имеет около 30.
Геном митохондрий имеет в своем распоряжении 22 гена для «собственных» транспортных РНК; 13 – для полипептидов, входящих в надмолекулярные комплексы, обеспечивающих дыхание органеллы; 2 гена для личных РНК.
Самое важное значение этой органеллы в том, что она вырабатывает АТФ. Проще говоря, является «электростанцией» в нашем организме, без нее клетки не могут полноценно функционировать; быстро «старятся» и погибают.
Что такое митохондриальный синдром?
При нарушении работы этих маленьких «энергоблоков» начинаются проблемы с энергообменом в клетке. При легких формах нарушения человек просто не выдерживает физических нагрузок, которые ему положено переносить по возрасту.
Однако более серьезные нарушения провоцируют необратимые изменения в энергообмене, и как следствие, сильные нарушения в работе клеток.
Митохондриальный синдром — это комплекс заболеваний, связанный с различными врожденными повреждениями митохондрий.
Причины синдрома
Такие органеллы как митохондрии делятся по-иному. Для них не присуще рекомбинирование генов, но при этом скорость мутации значительно выше. Во время деления митохондрии распределение генов между новыми клетками имеет совершенно случайный характер. Вероятность возникновение мутации от 1 до 99%. Причем спрогнозировать ее нет никакой возможности.
И чем больше больных генов, тем больше вероятность нарушения. Так как митохондрии наследуются по матери, то вследствие их мутирования в ее организме страдают дети обоих полов. Причем не избирательно, 1 или 2. Есть вероятность что все дети будут с аномалиями развития органов.
Мутации делят на два типа. Большинство белков «зашифровано» ядерной ДНК, которая также может видоизменяться по неясным причинам. Поэтому разделяют синдромы, вызванные как мутацией обычной митохондриальной кольцевой ДНК, так и ядерной.
Симптоматика
Определить четкий набор симптомов, присущих такой болезни, как митохондриальный синдром довольно сложно. Дело в том, что мутировавшие органеллы могут находиться в абсолютно любой клетке любого органа. И чем больше их накапливается, тем сильнее нарушается работа и этого органа, и всей системы, к которой он относится. В митохондрологии принято распределять синдромы в зависимости от вида пораженных тканей и от типа митохондриальной мутации.
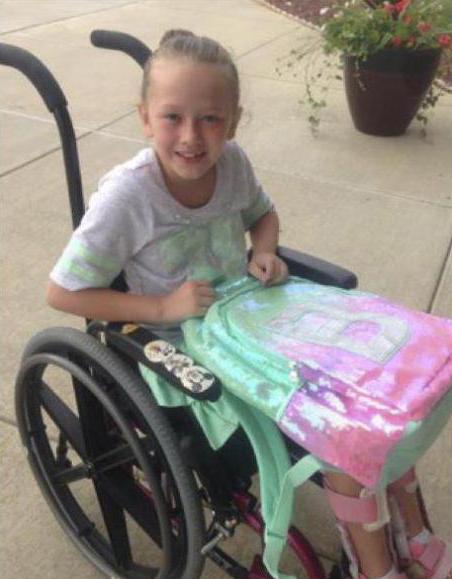
Обычно страдают те органы и системы, которые сильнее всего нуждаются в постоянной подаче кислорода – это мозг и центральная нервная система, печень, сердце, мышцы. Недополучая энергию, скелетные мышцы не поддерживают тело в вертикальном положении. В некоторых случаях появляются даже мышечные судороги.
Бывает, митохондрии настолько слабы в своей работе, что человек, получивший такой набор органелл от матери, полностью прикован к постели. При некоторых синдромах, о которых мы расскажем, человек страдает миоклонусами, гепатопатией, эпилептическими припадками и со временем деменцией, причем в совсем юном возрасте. Такие симптомы указывают на синдром митохондриального истощения.
Наиболее вероятные нарушения при мутациях митохондрий
Всего на сегодняшний день уже выявлено множество форм заболеваний, которые вызывает мутация митохондрий. Например, когда затрагивается мышечный каркас, диагностируют эпилептические приступы на фоне мышечного недоразвития. Причем мышечная структура не просто повреждена, она выглядит под микроскопом как недоразвитые волокна красного цвета. Мышечная атрофия в таком случае называется митохондриальной миопатией. Это наиболее частое нарушение при митохондриальной недостаточности. Если затронута именно сердечная мышца — кардиомиопатия, обнаружены патологические процессы в головном мозге — регистрируется энцефалопатия.
Что такое синдром митохондриальной энцефалопатии? Синдром диагностируют, когда есть нарушения в генах — tRNAs, MTND1, 4–6, MTCYB. При этом нарушается работа всей нервной системы.
Вместе с энцефалопатией наблюдаются и такой симптом, как лактат-ацидоз — или молочнокислая кома. Это осложнение, при котором молочная кислота начинает попадать в кровь.
Опасны и такие состояния у больных с синдромом митохондриальной недостаточности, как частые и злокачественные мигрени, у детей наблюдаются задержки психического и моторного развития, глухота, атаксия (проблемы с равновесием).
Симптомы не так хорошо изучены, поскольку заболевания, связанные с митохондриями не так давно открыты. Но об известных синдромах, клинические проявления которых пытаются лечить, мы расскажем.
Синдром МЕЛАС
МЕЛАС (MELAS) — энцефалопатия (проблемы с ЦНС), лактат-ацидоз, и в дополнение инсульты. Встречается синдром, как у младенцев, так и у взрослых людей. Но чаще симптомы начинают проявляться где-то с 5 до 15 лет. Что это за симптомы? Они перечислены в названии синдрома. У больного внезапно начинаются множественные инсульты — в височной и ли теменной зонах мозга. Присоединяются к инсультам и неврологические проблемы. Затем возникает мышечная слабость, сенсорная тугоухость. Возможны частые мышечные судороги.
Причиной синдрома считается подмена митохондриального гена в 3243-м положении. И лечение возможно только симптоматическое, то есть поддерживающая терапия.
Синдромы делеции митохондриальной ДНК
Начнем описание с такого заболевания, как синдром Кернса-Сейра, начинающийся с 4 лет. Синдром проявляется следующим образом:
- офтальмоплегия прогрессирующая;
- атаксия;
- атриовентрикулярная блокада сердца (замедление передачи импульса от одной сердечной камеры к другой);
- ретинит пигментный;
- те же красные рваные мышечные ткани.
Следующий синдром, имеющий те же «корни» — синдром Пирсона, который проявляется иначе:
- анемия гипопластическая, самый первый и опасный симптом;
- нарушение функций поджелудочной;
- позже возможны нарушения зрения;
- нарушения в костном мозге;
- появление деменции.
Синдром Пирсона обусловлен, как и синдром Кернса-Сейра, делециями митохондриальной ДНК. Делеции — это такие изменения в хромосомном наборе гена, при которых часть генного материала полностью утеряна.
Те аллели, которые мутировали, или потеряли части хромосом, не должны проявляться как доминантные. Но в митохондриальной ДНК все процессы хаотичны, мутирование происходит слишком быстро. Некоторые ученые даже считают, что митохондрии – это не органеллы, а бактерии, которые когда-то попали в человеческий организм и полностью прижились, создали симбиотическую связь с клеткой и начали служить ей. На такую теорию наталкивает тот факт, что у митохондрии свои, отдельные кольцевые ДНК.
Точковые мутации
К синдромам, обусловленным точковыми изменениями в материнской митохондрии относят синдром MERRF, NAPR, упомянутый MELAS и такое заболевание, как атрофия зрительного нерва Лебера.
Митохондриальный синдром MERRF — какие у него особенности?
- Наличествует атаксия — это нарушение координации, возможно, связанное с проблемами мозжечка. Человек плохо контролирует свои движения в пространстве.
- Симптомы миоклонической эпилепсии.
- Атрофия зрительного нерва (слепота от рождения) и глухота.
- Лактоацидоз.
- Нарушения чувствительности.
- Старт заболевания приходится на возраст от 3 лет.

Следующий вид заболевания NAPR— расшифровывается как невропатия, плюс атаксия, и плюс пигментный ретинит. При данном синдроме у ребенка прогрессируют нарушения в психомоторном развитии и деменция.
Синдром истощения ДНК
Синдром митохондриального истощения ДНК — весьма редкое заболевание. Ребенок с такой наследственной болезнью — инвалид с детства. Эти синдромы также подразделяются на множество видов.
Многие дети погибают от множественных дефектов развития внутренних органов, не дожив до 3 лет. Получение таких «покалеченных» митохондрий от матери происходит по аутосомно-рецессивному типу наследования. Генетики уверены, что в таких случаях имеют место множественные делеции.
Синдром также называют в научных кругах — синдром истощения митохондриальной ДНК. Заболевание проявляется у новорожденного сразу. У больного младенца наличествуют такие аномалии развития:
- Тяжелая гепатопатия — нарушение работы печени.
- Врожденная миопатия, выраженная в значительной слабости мышц.
- Кардиомиопатия — проблемы в работе сердечной мышцы.
- Атрофия мышц и отсутствие сухожильных рефлексов.
Основная причина таких заболеваний — это дефект межгеномной взаимосвязи (коммуникации).
Существует в генетике и такое понятие как синдром деплеции митохондриальной ДНК. Деплеция — это синоним истощения в генетике. При таком тяжелом синдроме генетический материал митохондрий истощен на 70–98%. Описан впервые не так давно, в 1991 году.
Что происходит с ребенком? В новорожденном периоде уже проявляется лактоацидоз, гипоальбуминемия (резкое снижение альбумина в крови), отеки и выраженная печеночная недостаточность. Наблюдались у некоторых больных и судороги. Симптом, который виден невооруженным взглядом — выраженная мышечная гипотония. Все дети, рожденные с такими признаками, не доживали и до года.
Причиной считается нарушение гена, который ответственен за репликацию ДНК. Его «неверная» работа приводит к тому, что практически все митохондрии мутируют и не выполняют свои функции. Тип наследования делеции митохондриальной ДНК может быть как аутосомно-рецессивным, так и аутосомно-доминантным.
Нарушения в ядерной ДНК
Кроме перечисленных митохондриальных синдромов есть другие, связанные с нарушениями в ядерной ДНК. Их тоже немало: Менкеса, Лея, Альперса, различные дефицитные состояния. Все они имеют прогрессирующее течение. Наиболее опасным считается синдром Лея, при котором ребенок практически не жизнеспособен уже с рождения.
Митохондриальный синдром у детей
Большинство заболеваний начинается с раннего детского возраста. В основном распространена миопатия, из-за которой дети не могут самостоятельно передвигаться и страдают мышечными болями. Кардиомиопатия — нарушения функции миокарда, также встречается довольно часто.
Митохондриальный синдром у ребенка, если недомогания не слишком серьезны и не угрожают здоровью, на протяжении всей жизни будет причинять беспокойства и мешать нормальному развитию. Таким детям нужны мероприятия по социализации. Им важно развивать скелетные мышцы, но не спортивными методами (так как у многих поражен миокард), а благодаря плаванию с дельфинами. Поэтому создан специальный фонд для таких детей, куда поступают деньги от благотворительности.

Одной из форм синдрома митохондриального истощения ДНК болеет мальчик по имени Чарли Гард, рожденный в 2016 году. Он с рождения не может самостоятельно глотать, пищу, дышать. Его состояние полностью контролируется врачами, и родители отчаянно борются за его жизнь. Хотя надежды мало. У него врожденная гепатопатия, он слеп и имеет тугоухость. Его родители надеются на современные методы лечения. Синдром получил также «народное» название — митохондриальный синдром Чарли.
Однако синдром митохондриального истощения ДНК однозначно приводит к летальному исходу. Врачи предупреждают об этом родителей сразу после установления диагноза. Множественные поражения органов и систем исключают нормальную жизнь для таких детей. Поэтому исключительно важно женщине перед беременностью пройти генетический анализ на мутации в митохондриях.
Тесты для диагностики
Диагностика подобных синдромов – это сложное задание для медиков. При постановке диагноза значение имеет комплексный анализ различных показателей. Проводится отдельно генетическое исследование, биохимическое, морфологическое, затем все данные сводятся воедино. Исследуется даже генеалогия ребенка.

Для точного медицинского заключения нужно провести также множество тестов для измерения различных соотношений. Например, проверяется пропорция в плазме крови лактат/пируват. Ведь недостаток пируватов и преобладание лактатов может означать начало лактоацидоза. Очень важно знать доктору о соотношении кетоновых тел в плазме. Но наиболее эффективным методом диагностики является биопсия мышц. Форму мутации можно узнать благодаря молекулярно-генетическому анализу ДНК.
Лечение синдромов
Трудность лечения заключается в отсутствии каких-нибудь механизмов, которые смогли бы заново «перестроить» мутировавшие гены. Врачи в таких случаях ничего не могут предпринять, кроме того, что назначить пируваты и некоторые витаминные комплексы. Особенно сложно помочь детям с множественными делециями генов. И если в карточке ребенка значится терминальная стадия синдрома митохондриального истощения ДНК, то это значит, что врачи полностью расписываются в своем бессилии.
Единственное, что может предложить медицина, это выявление митохондриальных мутаций у матери до беременности. Тогда можно попробовать пойти на экстракорпоральное зачатие, чтобы выносить здорового ребенка.
Источник
Митохондриальные болезни — это группа наследственной патологии, возникающей в результате нарушений клеточной энергетики, характеризующаяся полиморфизмом клинических проявлений, выражающаяся в преимущественном поражении центральной нервной системы и мышечной системы, а также других органов и систем организма [1].
Альтернативное определение митохондриальной патологии гласит, что это обширная группа патологических состояний, обусловленных генетическими, структурными и биохимическими дефектами митохондрий, нарушением тканевого дыхания и, как следствие, недостаточностью энергетического обмена.
Как указывает A. Munnich, «митохондриальные заболевания могут вызывать любой симптом, в любой ткани, в любом возрасте, при любом типе наследования» [2].
Митохондриальные дыхательные цепи — главный конечный путь аэробного метаболизма. Поэтому митохондриальную патологию нередко называют «болезнями дыхательной цепи митохондрий» (БДЦМ); это сравнительно новый класс болезней.
Исторические аспекты митохондриальной патологии
R. Luft и соавт. (1962) обнаружили взаимосвязь между мышечной слабостью и нарушениями процессов окислительного фосфорилирования в мышечной ткани [3]. S. Nass и M. Nass (1963) открыли существование собственного генетического аппарата митохондрий (обнаружены несколько копий кольцевой хромосомы) [4, 5]. В 1960–1970 гг. появилась концепция митохондриальных болезней, то есть патологии, этиологически опосредованной митохондриальной дисфункцией. В 1980-е гг. были получены точные молекулярно-генетические доказательства митохондриальной природы ряда заболеваний (болезнь Лебера, синдром Пирсона) [6].
Этиопатогенетические аспекты митохондриальной патологии
В зависимости от наличия основного метаболического дефекта принято рассматривать четыре основных группы митохондриальных болезней: 1) нарушения обмена пирувата; 2) дефекты обмена жирных кислот; 3) нарушения цикла Кребса; 4) дефекты электронного транспорта и окислительного фосфорилирования (OXPHOS) [1, 2].
Причинами возникновения митохондриальной патологии являются мутации в генах, кодирующих белки, задействованные в процессах энергообмена в клетках (включая субъединицы комплекса пируватдегидрогеназы, ферменты цикла Кребса, компоненты цепи транспорта электронов, структурные белки цепи транспорта электронов (ЦТЭ), митохондриальные транспортеры внутренней мембраны, регуляторы митохондриального нуклеотидного пула, а также факторы, взаимодействующие с ДНК митохондрий (мтДНК) [2, 6].
Митохондриальные нарушения связаны с большим числом болезней, не являющихся первичными митохондриальными цитопатиями. Тем не менее, при этих болезнях нарушения функций митохондрий вносят значимый вклад в патогенез и клинические проявления заболеваний. Описываемые болезни могут быть метаболическими, дегенеративными, воспалительными, врожденными/приобретенными мальформациями, а также неоплазмами.
Митохондрия является органеллой, которая присутствует практически в каждой клетке, за исключением зрелых эритроцитов. Именно поэтому митохондриальные болезни могут поражать любые системы и органы человеческого организма [6]. В связи с этим правильнее называть эти состояния «митохондриальными цитопатиями» [1, 2].
Основные особенности митохондриальных цитопатий включают выраженный полиморфизм клинических симптомов, мультисистемный характер поражения, вариабельность течения, прогрессирование и неадекватное реагирование на применяемую терапию.
Дыхательная цепь локализуется на внутренней мембране митохондрий и включает в себя пять мультиферментных комплексов, каждый из которых, в свою очередь, состоит из нескольких десятков субъединиц. Митохондриальная ДНК кодирует только 13 из белковых субъединиц дыхательной цепи, 2 белковых субъединицы мтРНК и 22 митохондриальных транспортных РНК (тРНК). Ядерный геном кодирует более 90% митохондриальных белков [2, 6].
Конечным результатом окислительного фосфорилирования, происходящего в комплексах 1-γ, является производство энергии (АТФ). Аденозин трифосфат — основной источник энергии для клеток.
Митохондриальная ДНК тесно взаимодействует с ядерной ДНК (яДНК). В каждом из 5 дыхательных комплексов основная часть субъединиц кодируется яДНК, а не мтДНК. Комплекс I состоит из 41 субъединицы, из которых 7 кодируются мтДНК, а остальные — яДНК. Комплекс II имеет всего 4 субъединицы; большая их часть кодируется яДНК. Комплекс III представлен десятью субъединицами; кодирование мтДНК — 1, яДНК — 9. Комплекс IV имеет 13 субъединиц, из которых 3 кодируются мтДНК, а 10 — яДНК. Комплекс V включает 12 субъединиц, кодирование мтДНК — 2, яДНК — 10 [2, 6].
Нарушения клеточной энергетики приводят к полисистемным заболеваниям. В первую очередь, страдают органы и ткани, являющиеся наиболее энергозависимыми: нервная система (энцефалопатии, полинейропатии), мышечная система (миопатии), сердце (кардиомиопатии), почки, печень, эндокринная система и другие органы и системы. До недавнего времени все эти заболевания определялись под многочисленными масками других нозологических форм патологии. К настоящему времени выявлено более 200 заболеваний, причиной которых являются мутации митохондриальной ДНК [1, 2, 6].
Митохондриальные болезни могут быть обусловлены патологией как митохондриального, так и ядерного генома. Как указывают P. F. Chinnery и соавт. (2004) и S. DiMauro (2004), мутации мтДНК были выявлены в 1 случае на 8000 населения, а распространенность митохондриальных заболеваний составляет порядка 11,5 случаев на 100 тысяч населения [7, 8].
В каждой клетке находятся от нескольких сотен до нескольких тысяч органелл — митохондрий, содержащих от 2 до 10 кольцевых молекул митохондриальной ДНК, способных к репликации, транскрипции и трансляции, причем независимо от ядерной ДНК.
Генетические аспекты митохондриальной патологии
Митохондриальная генетика отличается от классической менделевской в трех важнейших аспектах: 1) материнское наследование (всю цитоплазму, вместе с находящимися в ней органеллами, потомки получают вместе с яйцеклеткой); 2) гетероплазмия — одновременное существование в клетке нормального (дикого) и мутантного типов ДНК; 3) митотическая сегрегация (оба типа мтДНК в процессе деления клетки могут распределяться случайным образом между дочерними клетками) [1, 2].
Митохондриальная ДНК накапливает мутации более чем в 10 раз быстрее ядерного генома, так как она лишена защитных гистонов и ее окружение чрезвычайно богато реактивными видами кислорода, являющимися побочным продуктом метаболических процессов, протекающих в митохондриях. Пропорция мутантной мтДНК должна превышать критический пороговый уровень, прежде чем клетки начнут проявлять биохимические аномалии митохондриальных дыхательных цепей (пороговый эффект). Процентный уровень мутантной мтДНК может варьировать у индивидов внутри семей, а также в органах и тканях. В этом заключается одно из объяснений вариабельности клинической картины у больных с митохондриальными дисфункциями. Одни и те же мутации могут вызывать различные клинические синдромы (например, мутация A3243G — энцефалопатию с инсультоподобными пароксизмами — синдром MELAS, а также хроническую прогрессирующую наружную офтальмоплегию, сахарный диабет). Мутации в различных генах могут быть причиной одного и того же синдрома. Классическим примером такой ситуации является синдром MELAS [2].
Разновидности митохондриальной патологии
Если перечислить основные митохондриальные болезни, то в их числе окажутся следующие: митохондриальная нейрогастроинтестинальная энцефалопатия (MNGIE), синдром множественных делеций митохондриальной ДНК, липидная миопатия с нормальными уровнями карнитина, недостаточность карнитин пальмитоилтрансферазы, митохондриальный сахарный диабет, болезнь Альперса–Хуттенлохера, синдром Кернса–Сейра, болезнь Лебера (LHON), синдром Вольфрама, синдром MEMSA, синдром Пирсона, синдром SANDO, синдром MIRAS, синдром MELAS, синдром MERRF, синдром SCAE, синдром NARP, синдром Барта, синдром CPEO, синдром Ли и др. [1].
Наиболее часто в детском возрасте встречаются следующие клинические синдромы митохондриальной патологии: синдром MELAS (митохондриальная энцефаломиопатия, лактат-ацидоз и инсультоподобные пароксизмы), синдром MERRF (миоклонус-эпилепсия с рваными красными волокнами), синдром Кернса–Сейра (характеризуется птозом, офтальмоплегией, пигментным ретинитом, атаксией, нарушением сердечного проведения), синдром NARP (нейропатия, атаксия, пигментный ретинит), синдром Ли (подострая некротизирующая энцефаломиелопатия), болезнь Лебера (наследственная оптическая нейропатия) [1, 2].
Имеется большой пул заболеваний, причиной которых является не мутации митохондриальной ДНК, а мутации ядерной ДНК, кодирующей работу митохондрий. К ним относятся следующие виды патологии: болезнь Барта (миопатия, кардиомиопатия, транзиторные нейтро- и тромбоцитопении), митохондриальная гастроинтестинальная энцефалопатия (аутосомно-рецессивное мультисистемное заболевание): птоз, офтальмоплегия, периферическая нейропатия, гастроинтестинальная дисфункция, приводящая к кахексии, лейкоэнцефалопатия. Возраст дебюта последнего заболевания весьма вариабелен — от периода новорожденности до 43 лет.
Диагностика митохондриальной патологии
Клинические критерии диагностики митохондриальных болезней сравнительно многочисленны: 1) миопатический симптомокомплекс (непереносимость физических нагрузок, мышечная слабость, снижение мышечного тонуса); 2) судороги (миоклонические или мультифокальные); 3) мозжечковый синдром (атаксия, интенционный тремор); 4) поражение глазодвигательных нервов (птоз, наружная офтальмоплегия); 5) полинейропатия; 6) инсультоподобные пароксизмы; 7) мигренеподобные головные боли; 8) черепно-лицевая дисморфия; 9) дисметаболические проявления (рвота, эпизоды летаргии, комы); 10) дыхательные нарушения (апноэ, гипервентиляция, тахипноэ); 11) поражение сердца, печени, почек; 12) прогрессирующее течение заболевания [1, 2].
В диагностике митохондриальных болезней используются следующие клинические критерии: 1) признаки поражения соединительной ткани (гипермобильный синдром, гиперэластичность кожи, нарушения осанки и др.); 2) нейродегенеративные проявления, лейкопатии при проведении магнитно-резонансной томографии (МРТ) головного мозга; 3) повторные эпизоды нарушения сознания или необъяснимые эпизоды рвоты у новорожденных; 4) необъяснимая атаксия; 5) отставание в умственном развитии без определенных причин; 6) отягощенный семейный анамнез; 7) внезапное ухудшение состояния ребенка (судороги, рвота, расстройства дыхания, вялость, слабость, нарушения мышечного тонуса — чаще мышечная гипотония, кома, летаргия; поражение печени и почек, не поддающееся обычной терапии) [1, 2].
Лабораторные (биохимические) исследования нацелены в первую очередь на выявление у пациентов лактат-ацидоза и/или пируват-ацидоза. При этом следует помнить, что нормальные показатели молочной кислоты не исключают наличия митохондриального заболевания. Другие биохимические показатели, исследуемые при подозрении на наличие митохондриальной патологии, включают кетоновые тела в крови и моче, ацилкарнитины плазмы крови, а также содержание органических кислот и аминокислот в крови и моче [9].
M. V. Miles и соавт. (2008) предложили оценивать содержание мышечного коэнзима Q10 у детей с дефектом ферментов дыхательной цепи митохондрий [10].
Цитоморфоденситометрические исследования позволяют оценивать активность митохондрий лимфоцитов (снижение количества, увеличение объема, снижение активности).
Из инструментальных исследований (помимо методов нейровизуализации) используется биопсия скелетных мышц с проведением специфических гистохимических реакций — для выявления феномена «рваных красных волокон» (ragged red fibers — RRF) в полученном биоптате. Синдромами с «рваными красными волокнами» являются следующие: MELAS, MERRF, KSS, PEO (прогрессирующая наружная офтальмоплегия), а также синдром Пирсона. Синдромы без RRF: болезнь Leigh, NARP, LHON (наследственная оптическая нейропатия Лебера) [2].
Генетические методы исследований сводятся к определению наиболее частых мутаций и секвенированию митохондриальной ДНК.
Лечение митохондриальной патологии
Терапия митохондриальных болезней, к сожалению, не разработана. С позиций доказательной медицины считается, что эффективное лечение для этой представительной группы болезней отсутствует. Тем не менее, в различных странах мира используются фармакологические средства и биологически активные вещества, нацеленные на нормализацию метаболизма и обеспечение адекватной энергетики митохондрий.
При синдроме MELAS лечение должно быть направлено на лечение судорог, эндокринных расстройств, устранение последствий инсульта.
P. Каufmann и соавт. (2006) указывают, что поскольку уровень лактата часто коррелирует с тяжестью неврологических проявлений, целесообразно применять дихлорацетат для снижения уровня лактата [11]. В нашей стране с аналогичной целью используется диметилоксобутилфосфонилдиметилат (Димефосфон) [12].
В исследованиях японских авторов Y. Koga и соавт. (2002, 2005, 2006, 2007) с хорошим эффектом использовалось внутривенное введение L-аргинина (предшественника NO) — для стимуляции вазодилатации в остром периоде инсульта, а также пероральное его применение для снижения тяжести последующих эпизодов [13–16].
Среди средств, используемых в терапии митохондриальной патологии, фигурируют следующие: витамин В1 (тиамин) — 400 мг/сут, витамин В2 (рибофлавин) — 100 мг/сут, витамин С (аскорбиновая кислота) — до 1 г/сут, витамин Е (токоферол) — 400 МЕ/сут, никотинамид (ниацин) — до 500 мг/сут, коэнзим Q10 — от 90 до 200 мг/сут, L-карнитин — от 10 мг до 1–2 г/сут, янтарная кислота — от 25 мг до 1,5 г/cут, Димефосфон 15% — 1,0 мл на 5 кг массы тела. Применяются также цитохром С (внутривенно), Реамберин (внутривенно) и Цитофлавин (внутривенно и перорально) [17, 18].
В качестве других средств фармакотерапии выступают кортикостероиды, минералокортикоиды (при развитии надпочечниковой недостаточности), антиконвульсанты — при судорогах/эпилепсии (исключая вальпроевую кислоту и ее производные, ограничивая применение барбитуратов). В наших наблюдениях наиболее эффективной противосудорожной терапией являлось использование препаратов леветирацетам (Кеппра), топирамат (Топамакс) или их сочетаний.
Нейродиетология при митохондриальной патологии
Основным принципом диеты при митохондриальной патологии является ограничение нутриентов, оказывающих негативное влияние на механизмы обмена — до формирования метаболического блока (рацион питания одновременно обогащается другими компонентами на обычном или повышенном уровне). Такая терапевтическая стратегия получила название «обхождения блока» (going around the block). Важным исключением в этом плане является группа митохондриальных нарушений, ассоциированных с метаболизмом пирувата (недостаточность пируватдегидрогеназного комплекса с сопутствующими нарушениями со стороны углеводов/гликогена/аминокислот). При этом рекомендуются кетогенная диета и другие виды высокожировых диет [19].
Широко применяются вещества, являющиеся пищевыми кофакторами (коэнзим Q10, L-карнитин, ацетил-L-карнитин, витамин В2, аскорбиновая кислота, витамин Е, витамин В1, никотинамид, витамин В6, витамин В12, биотин, фолиевая кислота, витамин К, α-липоевая кислота, янтарная кислота, Se) [19]. Рекомендуется избегание индивидуальных алиментарных факторов, индуцирующих обострение митохондриальной болезни (голодание, потребление жиров, белков, сахарозы, крахмала, алкоголя, кофеина, мононатрия глутамата; количественные нарушения приема пищи и неадекватное потребление пищевой энергии). При необходимости осуществляется клиническое питание (энтеральное, парентеральное, гастростомия) [19].
Чрезвычайно важными являются своевременная диагностика митохондриальных болезней, поиск клинических и параклинических критериев этих заболеваний на этапе предварительном, догенетическом. Это необходимо для подбора адекватной метаболической терапии и предотвращения ухудшения состояния или инвалидизации больных с этими редкими заболеваниями.
C. S. Chi (2015) подчеркивает, что подтверждение или исключение митохондриальной патологии остается принципиальным в педиатрической практике, особенно когда клинические признаки болезни не являются специфичными, вследствие чего необходим катамнестический подход к оценке симптомов и биохимических показателей [20].
Литература
- Martikainen M. H., Chinnery P. F. Mitochondrial disease: mimics and chameleons // Pract. Neurol. 2015. Vol. 15 (6): 424–435.
- Sarnat H. B., Menkes J. H. Mitochondrial encephalomyopathies. Ch. 2. In: Child Neuroloy (Menkes J. H., Sarnat H. B., Maria B. L., eds). 7 th ed. Philadelphia-Baltimore. Lippincott Williams & Wilkins. 2006. 143–161.
- Luft R., Ikkos D., Palmieri G., Ernster L., Afzelius B. A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: a correlated clinical, biochemical, and morphological study // J. Clin. Invest. 1962. Vol. 41: 1776–1804.
- Nass M. M., Nass S. Intramitochondrial fibers with DNA characteristics. I. Fixation and electron staining reactions // J. Cell. Biol. 1963. Vol. 19: 593–611.
- Nass S., Nass M. M. Intramitochondrial fibers with DNA characteristics. II. Enzymatic and other hydrolytic treatments // J. Cell. Biol. 1963. Vol. 19: 613–629.
- Сухоруков В. С. Очерки митохондриальной патологии. М.: Медпрактика-М, 2011. 288 с.
- Chinnery P. F., DiMauro S., Shanske S., Schon E. A., Zeviani M., Mariotti C., Carrara F., Lombes A., Laforet P., Ogier H., Jaksch M., Lochmuller H., Horvath R., Deschauer M., Thorburn D. R., Bindoff L. A., Poulton J., Taylor R. W., Matthews J. N., Turnbull D. M. Risk of developing a mitochondrial DNA deletion disorder // Lancet. 2004. 364 (9434): 592–596.
- DiMauro S. Mitochondrial diseases // Biochim. Biophys. Acta. 2004. 1658 (1–2): 80–88.
- Siciliano G., Volpi L., Piazza S., Ricci G., Mancuso M., Murri L. Functional diagnostics in mitochondrial diseases // Biosci. Rep. 2007. Vol. 27 (1–3): 53–67.
- Miles M. V., Miles L., Tang P. H., Horn P. S., Steele P. E., DeGrauw A. J., Wong B. L., Bove K. E. Systematic evaluation of muscle coenzyme Q10 content in children with mitochondrial respiratory chain enzyme deficiencies // Mitochondrion. 2008. Vol. 8 (2): 170–180.
- Kaufmann P., Engelstad K., Wei Y., Jhung S., Sano M. C., Shungu D. C., Millar W. S., Hong X., Gooch C. L., Mao X., Pascual J. M., Hirano M., Stacpoole P. W., DiMauro S., De Vivo D. C. Dichloracetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial // Neurology. 2006. Vol. 66 (3): 324–330.
- Федеральное руководство по использованию лекарственных средств (формулярная система). Вып. XVI. М.: Эхо, 2015. 540.
- Koga Y., Ishibashi M., Ueki I., Yatsuga S., Fukiyama R., Akita Y., Matsuishi T. Effects of L-arginine on the acute phase of strokes in three patients with MELAS // Neurology. 2002. Vol. 58 (5): 827–828.
- Koga Y., Akita Y., Nishioka J., Yatsuga S., Povalko N., Tanabe Y., Fujimoto S., Matsuishi T. L-arginine improves the symptoms of strokelike episodes in MELAS // Neurology. 2005. Vol. 64 (4): 710–712.
- Koga Y., Akita Y., Junko N., Yatsuga S., Povalko N., Fukiyama R., Ishii M., Matsuishi T. Endothelial dysfunction in MELAS improved by L-arginine supplementation // Neurology. 2006. Vol. 66 (11): 1766–1769.
- Koga Y., Akita Y., Nishioka J., Yatsuga S., Povalko N., Katayama K., Matsuishi T. MELAS and L-arginine therapy // Mitochondrion. 2007. Vol. 7 (1–2): 133–139.
- Rai P. K., Russell O. M., Lightowlers R. N., Turnbull D. M. Potential compounds for the treatment of mitochondrial disease // Br. Med. Bull. 2015. Nov 20. pii: ldv046. [Epub ahead of print].
- Finsterer J., Bindu P. S. Therapeutic strategies for mitochondrial disorders // Pediatr. Neurol. 2015. Vol. 52 (3): 302–313.
- Студеникин В. М., Горюнова А. В., Грибакин С. Г., Журкова Н. В., Звонкова Н. Г., Ладодо К. С., Пак Л. А., Рославцева Е. А., Степакина Е. И., Студеникина Н. И., Турсунхужаева С. Ш., Шелковский В. И. Митохондриальные энцефалопатии. Глава 37. В кн.: Нейродиетология детского возраста (коллективная монография)/Под ред. Студеникина В. М. М.: Династия, 2012. С. 415–424.
- Chi C. S. Diagnostic approach in infants and children with mitochondrial diseases // Pediatr. Neonatol. 2015. Vol. 56 (1): 7–18.
В. М. Студеникин*, 1, доктор медицинских наук, профессор, академик РАЕ
О. В. Глоба**, кандидат медицинских наук
* ГОУ ВПО РНИМУ им. Н. И. Пирогова МЗ РФ, Москва
** ГОУ ВПО ПМГМУ им. И. М. Сеченова МЗ РФ, Москва
1 Контактная информация: studenikin@nczd.ru
Купить номер с этой статьей в pdf
Источник