
Мкб 10 код миодистрофия

Медицинский эксперт статьи
х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Заболевание, которое называется дистрофия Дюшена, связывают с повреждением генных структур, ответственных за продукцию крупного мышечного белка дистрофина. Такая патология передается наследственным путем, аутосомно-рецессивным типом наследования: то есть, данная патология как бы прячется, либо проявляется через поколение. Данный тип сцеплен с X-хромосомой.
[1]
Причины дистрофии Дюшена
Патология появляется вследствие генной мутации, имеющей место в области хр21. Более четверти таких патологий связывают со стойким изменением генотипа в материнской яйцеклетке. Остальные случаи объясняют гетерозиготностью мамы пациента по патологии мутагенеза в гене дистрофина.
Принято считать, что примерно 7% всех периодически возникающих случаев заболевания – это следствие образования в женском яичнике нескольких клеточных генераций с мутированными и обычными аллеями дистрофина. При этом наиболее распространенным типом мутации (около 65%) являются значительные потери участков хромосомы. У 5% пациентов обнаруживают удвоение участка хромосомы, а в оставшихся случаях патологии – point mutation, когда затрагивается один или несколько нуклеотидов, тогда как к мутации относятся более протяженные дефекты гена.
Патология передается по аутосомно-рецессивному типу, со сцепкой с X-хромосомой (поражает мужские особи). Больше половины таких патологий возникают спонтанно, в связи с мутацией генов.
При генетическом обследовании большую роль играет определение у сестер пациента скрытых признаков заболевания. От такой носительницы мутированного гена может произойти передача патологии 50% её детей мужского пола, а 50% её дочек станут такими же носителями мутированного гена.
Женщины, которые обладают поврежденным геном, делятся им со своим ребенком, хотя сами миопатией не страдают. В основном заболеванию подвержены мальчики. Девочки также могут заболеть, но это случается крайне редко: такое может произойти лишь при дефекте структуры хромосом.
[2]
Симптомы дистрофии Дюшена
Начальные симптомы дистрофии Дюшена можно заметить уже в возрасте от 1 до 5 лет. Для больного ребенка характерно торможение ранней двигательной активности. При попытках ходить самостоятельно (у детей, старше 1 года) можно наблюдать постоянные падения, запутывание ножек, быструю усталость. Если малыш все-таки начинает ходить, то при этом он переваливается с ноги на ногу (утиная походка), ему сложно подниматься по ступенькам и вставать с колен.
Постепенно у маленьких пациентов возникает увеличение объемов различных мышечных групп, что внешне похоже на сильно накачанные мышцы. С дальнейшим течением и усугублением патологии такое увеличение переходит, наоборот, в уменьшение мышц.
Заболевание распространяется по телу восходящим путем: от мышц ног и малого таза до спины, плеч и рук.
Уже на начальных этапах заболевания можно наблюдать понижение сухожильных рефлексов. Далее развивается искривление позвоночника, грудная клетка становится седло- или килевидной, деформируются стопы. Возникают проблемы с сердечной мышцей: появляются признаки нарушения сердечного ритма и левожелудочковая гипертрофия. У четверти пациентов обнаруживают симптомы умственной отсталости: чаще всего это проявляется признаками олигофрении.
Приблизительно к 12-летнему возрасту пациенты перестают ходить, через 2-3 года полностью теряют способность совершать движения. В 20-30 лет большее количество таких больных умирает. На поздних этапах заболевания мышечная слабость переходит и на дыхательную и глотательную систему. Летальный исход наступает от присоединившихся бактериальных инфекций или от недостатка дыхательной и сердечной деятельности.
[3], [4], [5], [6]
Формы
[7], [8], [9], [10]
Мышечная дистрофия Дюшена
Мышечная дистрофия Дюшена, к счастью, встречается относительно редко и проявляется в мышечной слабости. Согласно статистике, такая патология встречается приблизительно у одного малыша из 3000 новорожденных детей. Кроме того, известны несколько достаточно редких форм миопатии, отличающиеся менее выраженными проявлениями.
Развитие мышечной дистрофии связано с медленным разрушением соединений между нервными и мышечными волокнами.
Девочки, родившиеся от матери с поврежденным геном, также могут стать носителем такого повреждения, хотя заболевание у них практически никогда не проявляется.
Кроме дистрофии Дюшена, медицина выделяет ещё и другие виды миопатий, встречающиеся крайне редко:
- синдром Беккера (также поражает мальчиков, имеет врожденный тип, но проявляет себя только к периоду полового созревания и отступает приблизительно к 45 годам);
- врожденная форма миопатии (поражает малышей независимо от пола, но может встречаться, можно сказать, лишь в единичных случаях);
- лопаточно-плече-лицевая форма миопатии – проявляется не сразу, а на протяжении около 10 лет. При патологии отмечается слабость мимических мышц, вялая реакция мускулатуры лица при попытке выразить определенные эмоции;
- патология Эмери-Дрейфуса (подобный вид миопатии с негативными последствиями для миокарда).
[11], [12], [13], [14], [15]
Прогрессирующая дистрофия Дюшена
Прогрессирующая дистрофия Дюшена – это серьезная, наиболее распространенная форма миопатии. Развивается она в младенческом возрасте, обычно у детей до 3-х лет, изредка в более старшем возрасте. Практически у всех пациентов (за небольшим исключением) наблюдается увеличение размеров икроножных мышц (иногда в совокупности с дельтовидными и четырехглавыми мышцами). Такое увеличение часто связано с жировой инфильтрацией мышц, однако в некоторых случаях в действительности увеличиваются именно мышцы.
Уменьшение объема мышечной массы наблюдается в основном в области спины и тазового пояса. Наряду с атрофическими расстройствами зачастую можно отмечать наличие умственной отсталости.
Не редкостью являются нарушения целостности и формы костей даже вследствие незначительных нагрузок и травм. Спустя 5-10 лет от первых проявлений патологии можно обнаружить поражение сердечной мышцы, что выражается тахикардией и нарушениями ЭКГ. Характерным признаком считается усиленная активность сывороточной креатинкиназы.
В целом, заболевание протекает тяжелее, чем при остальных формах миопатии. Атрофические изменения быстро распространяются по всему организму. Большая часть пациентов к 10-летнему возрасту практически лишена возможности двигаться. Такие больные крайне редко способны дожить до 30 лет, погибая от сопутствующих заболеваний.
Диагностика дистрофии Дюшена
Диагноз дистрофии Дюшена должен быть подтвержден методом генетического тестирования, однако в некоторых случаях возможно назначение других исследований.
- Генетический тест проводится однозначно, даже если доктор уверен в том, что у пациента именно мышечная дистрофия. При помощи данного метода можно определиться с четкими характеристиками патологических нарушений в ДНК. Помимо постановки диагноза, это исследование поможет родителям решить вопрос о будущих беременностях. Также результаты генетического теста будут полезны родственницам матери-носительницы мутированного гена.
- Доктор может посоветовать пройти биопсию мышечных волокон. Такое исследование покажет, продуцируется ли в организме белок дистрофин, и если продуцируется, то в каком количестве. При помощи биопсии специалисты определяют точное количество белка в миоцитах. Но биопсия не может быть заменой генетическому анализу!
- Метод электромиографии (определение проводимости нервных импульсов) был актуален ещё несколько лет назад, однако сейчас он необязателен.
- Исследование крови на креатинкиназу: при дистрофии Дюшена количество данного энзима значительно превышает норму.
- Оценка сердечной деятельности, дыхательной системы, мышечных возможностей, проведение ЭКГ, определение сердечных биологических маркеров и плотности костной ткани.
Очень важно определить точный диагноз, если специалисты подозревают у ребенка миопатию. Причем сделать это необходимо в самом скором времени. Квалифицированное лечение доктор назначит на основании этих исследований, предварительно проведя беседу с родителями и разъяснив им все особенности заболевания.
[16], [17], [18], [19]
Лечение дистрофии Дюшена
В настоящее время лекарства от дистрофии Дюшена ещё не изобрели. Хотя научные исследования на эту тему проводятся достаточно интенсивно: над этим работают ученые Великобритании, Израиля и США. Новейшие методы, находящиеся в стадии разработки:
- пропуск экзонов – это процедура, способствующая замедлению скорости нарастания миопатии. Этот метод смягчает течение заболевания, значительно облегчает симптомы, но не устраняет мутацию;
- введение гена дистрофина с применением вирусных переносчиков генов или плазмид – позволяет пациентам более длительно сохранять способность к движениям и ходьбе, что в большой степени улучшает качество их жизни;
- трансплантация миогенных клеток – это введение фибробластов, что способствует усилению синтеза нового немутированного дистрофина. Данный метод имеет ряд преимуществ: это длительный положительный результат, возможность сочетать процедуру с другими лечебными методами, возможность использования практически в любом возрасте и контролированная продукция нового дистрофина;
- восстановление мышечных волокон с применением эмбриональных стволовых клеток, мышечных стволовых клеток – данные методы улучшают регенерацию мышц, позволяют дистрофину продуцироваться в больших количествах, укрепляют мышечную структуру и значительно восстанавливают функции мышц;
- регуляция атрофина для замещения дистрофина – метод, основанный на эксперименте, доказывающем, что при недостатке атрофина наблюдаются такие же симптомы, как и при недостатке дистрофина. Эти белки похожи по строению и функциям. Путем длительных научных исследований ученые пришли к выводу, что регуляцию гена атрофина можно применять в качестве лечения дистрофии Дюшена;
- блокирование миостатина. Миостатин – это неактивный белок, который обладает способностью запускать биохимические процессы, тормозящие формирование мышц. Соответственно, блокирование этого белка должно содействовать росту мышечной ткани;
- блокирование трансформирующего фактора роста β – белка, тормозящего функцию миосаттелиоцитов (миогенных стволовых клеток). Такой метод поможет уменьшить степень фиброза;
- повышение регуляции инсулиноподобного фактора роста-1 – белка, сходного по структуре с инсулином. Фактор роста-1 повышает качество мышечной ткани, активирует развитие и увеличивает силу мышц.
На данный момент специалисты предлагают следующее лечение дистрофии Дюшена:
- прием кортикостероидных препаратов для увеличения мышечной силы и облегчения состояния пациента;
- применение β-2-агонистов для временного придания силы мышцам;
- физиотерапевтические процедуры, миостимуляция;
- ортопедическая помощь (коляски, ходунки, фиксаторы голени и пр.).
«Эликсира исцеления» от дистрофии Дюшена, к сожалению, не существует, поэтому при поисках эффективного лечения с крайней осторожностью относитесь к препаратам и процедурам, которые могут быть представлены вашему вниманию как «панацея».
Не покупайте неизвестное лекарство, если нет достоверных подтверждений его эффективности. Помните, что вы можете потратить большую финансовую сумму, и, к тому же, не только не помочь, но и навредить своему малышу.
Профилактика
Конечно, говорить о профилактике наследственного заболевания, связанного с мутацией гена, сложно. Безусловно, в семьях, в которых имеются случаи рождения детей с мышечной дистрофией Дюшена, в обязательном порядке необходимо проводить консультации генетика, желательно ещё до начала планирования беременности молодой парой.
Если ребенок с патологией мышц уже появился на свет, то необходимо приложить все усилия, чтобы не допустить потерю мышечной ткани и как можно дольше поддерживать у ребенка двигательную активность. В таких ситуациях недопустимо опускать руки и позволять мышцам атрофироваться. Следует заниматься с малышом специальной гимнастикой, чтобы сохранить амплитуду движений в суставах. Для предупреждения контрактур рекомендуется применять поддерживающие корсеты и фиксаторы.
Дети, которые хоть немного, но могут ходить, должны это делать как можно чаще. Не загоняйте ребенка в постель: пусть будет активным, это позволит мышцам замедлить регрессию. Таким детям необходимо уделять максимум внимания и заботы, они не должны чувствовать себя обделенными или обиженными.
Хороший эффект дает плавание, которым заниматься можно и даже нужно. Помните: ничегонеделание (постоянный постельный режим) лишь ускорит прогрессирование болезни. Необходимо позволять больному, по крайней мере, самому удовлетворять свои потребности.
[20], [21], [22], [23], [24], [25]
Прогноз
Дистрофические процессы при заболевании поражают всю мышечную систему: мышцы дыхательной системы, сердца, скелета. Пациенты с дистрофией Дюшена чаще всего доживают до 15, максимум до 30 лет. Исследования, проводимые мировыми учеными в данном направлении, дают надежду на улучшение качества и продолжительности жизни таких пациентов.
И сейчас известны случаи, когда больные могли дожить до 40 или даже 50 лет. Такой результат был обусловлен наличием специального и постоянного ухода: оборудования для поддержки дыхательной системы, адекватной лекарственной терапии.
Обнаружение гена, который является главным фактором развития болезни, дало огромный толчок к продолжению научных экспериментов в терапии генных мутаций. Однако, на данный момент, к сожалению, не существует какого-либо лечения, которое помогло бы абсолютно вылечить миопатию. Используемые методы лишь дают возможность улучшить и продлить жизнь больного.
Остается верить, что в скором времени наука найдет способ исправления наследственной патологии, и дистрофия Дюшена, наконец, будет побеждена.
[26]
Источник
Связанные заболевания и их лечение
Описания заболеваний
Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Дифференциальная диагностика
- Лечение
Названия
Название: Прогрессирующая мышечная дистрофия Беккера.
Прогрессирующая мышечная дистрофия Беккера
Описание
Прогрессирующая мышечная дистрофия Беккера. Вариант наследственной сцепленной с Х – хромосомой миодистрофии, отличающейся более замедленным и доброкачественным течением. Заболевание характеризуется постепенно усугубляющейся и распространяющейся мышечной слабостью, гипотонией и атрофией, первоначально возникающей в мышцах бедер и тазового пояса. Диагностический поиск включает неврологическое обследование, консультацию генетика и кардиолога, нейрофизиологическое тестирование нервно-мышечного аппарата, ДНК диагностику, биопсию мышц с морфологическим, иммунологическим и гистохимическим изучением полученных образцов. Лечение симптоматическое и, к сожалению, малоэффективное. Прогрессирование болезни приводит к потери больными способности самостоятельно передвигаться к возрасту 40 лет.
Дополнительные факты
Прогрессирующая мышечная дистрофия Беккера впервые была описана в 1955 г. Как доброкачественный вариант течения мышечной дистрофии Дюшенна. В последующем многочисленные исследования в области неврологии, генетики и биохимии обнаружили существенные отличия в характере течения, биохимической и морфологической основе этих заболеваний. В результате клиническая форма Беккера была выделена как самостоятельная нозология.
Мышечная дистрофия Беккера входит в группу миопатий (миодистрофий) — заболеваний, возникающих вследствие нарушений строения и метаболизма мышечной ткани и проявляющихся мышечной слабостью. Патология наследуется рецессивно сцеплено с Х-хромосомой, поэтому болеют только лица мужского пола. Частота встречаемости составляет 1 новорожденный на 20 тыс. Детей.
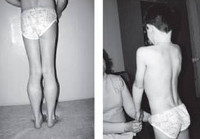
Прогрессирующая мышечная дистрофия Беккера
Причины
В основе заболевания лежит мутация в гене, ответственном за кодирование белка дистрофина. Примерно 30% от общего числа случаев мышечной дистрофии Беккера приходится на т. Н. «свежие» мутации. Ген располагается в 21 локусе (в регионе Хр21. 2–р21. 1) короткого плеча Х-хромосомы. Примерно у 65-70% больных обнаруживаются крупные делеции указанного участка, у 5% – дупликации, у остальных — точковые мутации. Указанные структурные перестройки гена не влекут за собой полного прекращения синтеза дистрофина, как при дистрофии Дюшенна, а потенцируют синтез аномального усеченного белка, в некоторой степени способного выполнять свои функции. Это и обуславливает более доброкачественный характер дистрофии Беккера в сравнении с вариантом Дюшенна.
В норме белок дистрофин поддерживает целостность сарколеммы – мембраны миоцитов (мышечных волокон), обеспечивает эластичность и устойчивость миофибрилл при мышечном сокращении. Неспособность аномального дистрофина адекватно выполнять эти функции приводит к нарушению целостности мембран мышечных волокон. В следствие этого происходят дегенеративные изменения цитоплазматических компонентов последних и повышенная транспортировка ионов калия внутрь миоцитов. Результатом таких биохимических и морфологических сдвигов является гибель миофибрилл и разрушение мышечных волокон. На месте погибших миоцитов происходит образование соединительной ткани, что обуславливает феномен псевдогипертрофии — увеличение объема и плотности мышцы при резком снижении ее сократительной способности.
Симптомы
Прогрессирующая мышечная дистрофия Беккера манифестирует обычно в период от 10 до 15 лет, в некоторых случаях раньше. Начальными признаками заболевания выступают чрезмерная утомляемость и мышечная слабость в тазовом поясе и нижних конечностях. У ряда пациентов первыми проявлениями являются периодические болезненные мышечные судороги (крампи), локализующиеся в ногах. Мышечная слабость обуславливает затруднение при подъеме по лестнице, при необходимости встать из положения сидя. Со временем формируется переваливающаяся «утиная» походка. Для того, чтобы встать, пациент вынужден использовать вспомогательные миопатические приемы — опираться руками о расположенные рядом предметы мебели или, при отсутствии таковых, использовать в качестве опоры собственное тело (симптом Говерса).
Как и другие наследственные миопатии, заболевание Беккера характеризуется симметрично развивающимися атрофиями мышц. В первую очередь поражаются мышцы бедра и тазового пояса, затем процесс распространяется на мускулатуру плечевого пояса и проксимальных мышц рук. В начале болезни формируются псевдогипертрофии, наиболее выраженные в икроножных, дельтовидных, трех- и четырехглавых мышцах. По мере прогрессирования миодистрофии они трансформируются в мышечные гипотрофии.
Ассоциированные симптомы: Слабость в руках. Слабость мышц (парез). Судороги.
Диагностика
Прогрессирующая мышечная дистрофия Беккера диагностируется неврологом на основании анамнеза, клинических данных, дополнительных обследований и генетического тестирования. В неврологическом статусе наблюдается снижение мышечной силы и умеренное снижение мышечного тонуса в проксимальных отделах конечностей, выпадение коленных рефлексов при симметричном снижении сухожильных рефлексов дистальных отделов ног и верхних конечностей, полная сохранность чувствительности.
Среди клинических анализов наибольшее значение имеет биохимический анализ крови, который выявляет многократное повышение уровня КФК. Данные электронейрографии позволяют исключить поражение нервных волокон, электромиография свидетельствует о первично-мышечном типе поражения. Биопсия мышц проводится только после отрицательных результатов генетического анализа. Морфологическое исследование полученного материала определяет диффузную разнокалиберность, дистрофические и некротические изменения мышечных волокон, разрастание соединительной ткани. Проводится специальное иммунное окрашивание образцов с последующим определением наличия в них дистрофина.
Подтвердить диагноз мышечной дистрофии Беккера позволяет консультация генетика с проведением анализа ДНК. Выявление дупликаций или делеций в гене Хр21 дает возможность установить точный диагноз. Отрицательный результат анализа ДНК не говорит об отсутствии патологии, поскольку могут иметь место точковые мутации, поиск которых представляет собой сложную и более дорогостоящую процедуру.
С целью выявления сердечной патологии назначается электрокардиография, Эхо-КГ, консультация кардиолога. Кардиологическое обследование может обнаружить нарушение внутрижелудочковой проводимости, АВ-блокаду, дилатацию желудочков, гипертрофические изменения миокарда, кардиомиопатию, сердечную недостаточность.
Дифференциальная диагностика
Дифференциальная диагностика проводится с прогрессирующей мышечной дистрофией Дрейфуса, миодистрофией Дюшена, мышечной дистрофией Эрба-Рота, метаболической миопатией, полимиозитом и дерматомиозитом, воспалительной миопатией, спинальной амиотрофией, наследственной полиневропатией.
Пренатальная диагностика рекомендована, когда мать является носителем патогенного гена. Если ребенок мужского пола, то вероятность развития заболевания у него составляет 50%. Биопсия хориона может проводиться в сроке 11-14 нед. Беременности, амниоцентез — после 15-й недели, забор пуповинной крови (кордоцентез) — на сроке больше 18 нед.
Лечение
На современном этапе несколькими группами ученых ведутся настойчивые исследования в области поиска эффективных методов лечения прогрессирующих миодистрофий. В настоящее время пациенты получают в основном метаболическую и симптоматическую терапию. Разработаны различные схемы лечения, позволяющие улучшить двигательные возможности больного и несколько замедлить прогрессирование болезни. Пациентам назначают актопротекторы (этилтиобензимидазол), неостигмин, АТФ, анаболические стероиды (метиландростендиол), сердечные средства. По вопросу длительной терапии глюкокортикоидами (преднизолоном) клиницисты имеют различные мнения. Одни считают, что подобное лечение тормозит прогрессирование миодистрофии, другие отвергают это предположение.
Наблюдения показали, что постельный режим усугубляет мышечную слабость. Поэтому пациентам рекомендуется умеренная физическая активность, занятия плаваньем. Поддержание мышечной эластичности и силы, а также профилактика контрактур проводится средствами массажа, физиотерапии и лечебной гимнастики. По показаниям проводится хирургическое лечение контрактур. Применение различных ортопедических средств (ходунков, инвалидных колясок, фиксаторов для ног, экзоскелетов) позволяет расширить двигательные возможности пациентов и их способность к самообслуживанию. По показаниям проводится хирургическое лечение контрактур.
Источник