
Мукополисахаридоз 1 типа синдром гурлера что это

Мукополисахаридоз – общее название ряда редких заболеваний, которые носят генетический характер. Патология развивается вследствие нехватки в организме некоторых ферментов, помогающих расщеплять жиры и углеводы на простые молекулы. В данной статье рассмотрен мукополисахаридоз 1 типа – синдром Гурлера.
Причины
Болезнь является наследственной по аутосомно-рецессивному типу. Она развивается по причине аномалий обмена мукополисахаридов.
Патогенез
Мукополисахаридоз относится к так называемым лизосомным болезням накопления. В результате дефицита лизосомных ферментов затрудняется катаболизм гликозаминогликанов. Они накапливаются в тканях и органах, нарушая работу организма и его систем. В первую очередь происходит поражение скелета и задержка физического развития.
Внешние признаки и симптомы заболевания
Симптомы заболевания проявляются в виде дефектов костной, соединительной, хрящевой тканей. Основной признак – задержка роста. Этот симптом можно обнаружить раньше остальных, обычно уже к концу первого года жизни становится понятно, что ребенок отстает в росте.
Также мукополисахаридоз можно предположить, видя грубые черты лица. У больных большой язык, гипертелозирм (слишком большое расстояние между парными органами, в данном случае между глазами), деформированные ушные раковины, лоб нависает, зубы искривлены.

К симптомам мукополисахаридоза относятся деформации грудной клетки, выраженный кифоз грудопоясничного отдела позвоночника. При проведении рентгена можно обнаружить преждевременное окостенение затылочно-теменного шва без нарушения ядер окостенения.
В большинстве случаев болезнь сопровождает ограничение подвижности суставов, абдоминальные грыжи, гепатоспленомегалия (увеличение печени и селезенки из-за патологических процессов, происходящих в результате заболевания).
Со стороны неврологии отмечаются двигательная заторможенности и мышечная гипотония. Также при мукополисахаридозе происходит ослабление слуха и снижение интеллекта вплоть до тяжелого слабоумия. Вследствие прогрессирующих системных поражений скелета внутренние органы также подвергаются нарушениям в различной степени.
Типы мукополисахаридоза
Различают несколько типов заболевания, которые различаются выраженностью костных изменений и нарушений психики:
- I – синдром Гурлера.
- II – синдром Гунтера (Хантера).
- III – синдром Санфилиппо.
- IV – синдром Моркио.
- VI – синдром Марото – Лами.
- VII – синдром Слая.
Деление в медицинских практиках разных стран может отличаться. В тип V обычно выделяют синдром Шейе. В американском же сообществе больных мукополисаридозами делят по тяжести проявления симптоматики первого типа и выделяют три фенотипа: синдром Гурлера, синдром Шейе и промежуточный между ними синдром Гурлер – Шейе (из них Гурлер самый тяжелый, Шейе – самый легкий).
Синдром Гурлера
Эта форма встречается чаще остальных и была описана ранее других синдромов. К тому же клиническая картина наиболее яркая и типичная из всех видов мукополисахаридоза.
Синдром Гурлера развивается в результате аутосомно-рецессивного наследования. Этот тип болезни характеризуется очень быстрым прогрессом. Несмотря на то что мукополисахаридоз первого типа схож со вторым (Гунтера, или Хантера), это более сложное заболевание. Впервые данная форма была описана в 1919 году Гертрудой Гурлер (поэтому правильное название – синдром Гурлер, а не Гурлера). Частота появления – один случай на 20-25 тысяч человек, причем в большинстве случаев родители заболевшего ребенка находятся в кровном родстве. Поэтому если ставится диагноз “синдром Гурлера”, причины необходимо искать на генетическом уровне. Симптомы проявляются практически сразу после рождения, а к году-двум клиническая картина уже выражена полностью.
Синдром Гурлера – классическое проявление заболевания. По мере развития недуга замедляется рост, отмечается видимое помутнение роговицы, сосуды носа переполняются кровью. При этой форме заболевания при рентгенологическом исследовании можно обнаружить расширение турецкого седла, укорочение и расширение длинных костей, гипоплазию и заостренность позвонков поясничной области (так называемые рыбьи позвонки), деформации позвоночного столба (больные страдают кифозом и лордозом грудопоясничной области позвоночника). Начинаются патологии сердечно-сосудистой системы – закупориваются коронарные артерии, изменяются клапаны, миокард, эндокард, сердце увеличивается в размерах.
Отмечается гидроцефалия, причиной которой становятся отложения мукополисахаридов в мозговых оболочках. Определяются очаги демиелинизации. Мукополисахариды также откладываются в печени, селезенке, эпителии почечных канальцев; сетчатке, склере, роговице глаз; нервных клетках, хрящах.
Дети рождаются уже с характерным внешним видом – у них очень своеобразные, грубые черты лица, из-за чего другое название мукополисахароидозов – гаргоилизм (от слова «гаргулья» – фантастическая фигура с необычными чертами лица), в том числе так называют и синдром Гурлера. Фото, на которых изображены больные, наглядно иллюстрируют причудливое искажение черт лица ребенка. У таких детей изменен череп – он приобретает форму киля лодки, наблюдается так называемая скафоцефалия, запавшая переносица, толстые губы, большой язык, крутой лоб, короткая шея и характерное выражение лица. Внешне это действительно напоминает то, как изображают мифологических гаргулий.

Также у них укорочена грудная клетка, нижние ребра выступают, есть признаки кифоза, суставы (особенно пальцев и локтей) малоподвижны, могут быть паховые и пупочные грыжи. Ногти могут приобретать вид часовых стекол, волосы становятся жесткими и сухими, голос – низким и хриплым. Вероятна тугоухость или даже глухота. Больные часто страдают от кариеса, который провоцирует синдром Гурлера.
Симптомы включают патологии дыхательной системы, из-за этого ребенок дышит ртом, у него появляются аденоиды, он подвержен вирусным инфекциям. Со временем у него развиваются характерные для мукополисахаридозов проблемы с печенью и селезенкой (в результате увеличен живот), слабоумие.
Рост остается карликовым. Из-за неправильного телосложения и деформации позвоночника больные ходят на полусогнутых ногах, на цыпочках.
У синдрома Гурлера злокачественный прогрессирующий характер, поэтому крайне быстро наступает инвалидизация больных. Многие не доживают даже до 10 лет.
Диагностика
Пациенту необходимо провести клиническое, рентгенологическое, биохимическое, генеалогическое, а также молекулярно-генетические исследования. Диагностика проводится по клиническим проявлениям болезни, на основе рентгенологических исследований и анализа мочи, по которому определяется активность ферментов и экскреция гликозаминогликанов.
Лечение мукополисахаридоза
Если больному поставлен диагноз “синдром Гурлера”, лечение предполагается больше симптоматическое. Пациент наблюдается комплексно у ортопеда, хирурга, педиатра, отоларинголога, нейрохирурга, офтальмолога и невропатолога. Больному проводится ортопедическая коррекция нарушений опорно-двигательного аппарата, удаляют грыжи, лечатся часто возникающие у таких пациентов вирусные заболевания, нарушения слуха, отиты, синуситы. Также под наблюдение попадает сердечно-сосудистая система.
Используются гормональные препараты, которые временно улучшают состояние пациента:
- глюкокортикоиды,
- кортикотропин,
- тиреоидин.
Помимо этого больному показаны витамин А, декстран 70, которые также временно улучшают состояние больного. Недолгосрочное улучшение дает переливание препаратов плазмы крови.
При синдроме Гурлера пациенту может быть назначено физиотерапевтическое лечение: электрофорез лидазы на область пораженных суставов, лазерная пунктура, магнитотерапия, парафиновые аппликации. Также больным рекомендуется заниматься лечебной физкультурой, упражнения которой воздействуют на суставы и позвоночник. Хорошие результаты часто дает массаж.
Так как больные синдромом Гурлера подвержены респираторным заболеваниям, необходимо своевременно проводить санацию очагов инфекций во рту и носоглотке.
Для лечения мукополисахаридоза 1-го типа часто проводятся хирургические вмешательства – пересадка роговицы и коррекция клапанных пороков сердца и ущемлений нервов. В международной практике помимо симптоматического лечения медикаментами, физиотерапией или хирургических вмешательств используется заместительная ферментная терапия, а также трансплантация стволовых клеток.
При необходимости больному проводят грыжеиссечения, удаление аденоидов, антиглаукоматозные операции, трахеостомию, протезирование тазобедренных суставов, шунтирование при гидроцефалии и пр.
Прогноз
Прогноз неблагоприятен как для синдрома Гурлера, так и для других форм, которые имеет мукополисахаридоз. Синдром Гурлера отличается наибольшей безнадежностью. Изменения скелета с каждым годом увеличиваются, в результате органы и системы подвергаются более значительным нарушениям. Если ребенок не умирает от пневмонии в раннем возрасте, то к 7-12 годам это уже беспомощный физически и психически инвалид. До юношеского возраста доживают единицы.
Профилактика
Предупредить данное заболевание невозможно. Но можно обнаружить его на самой ранней стадии – при пренатальной диагностике. Для этого проводится анализ амниотических клеток на предмет дефицита фермента (в случае положительного результата беременной рекомендуется аборт).
За счет ранней диагностики и своевременной терапии развившейся компрессии спинного мозга можно избежать необратимых повреждений нервов. Для профилактики обязательно проводится медико-генетическая консультация.
Прогнозы лечения
Несмотря на сложности в плане лечения синдрома Гурлера, в последние 20 лет во многих развитых странах используется трансплантация костного мозга, которая значительно улучшает качество жизни больных. Более 10 лет применяются препараты заместительной терапии для лечения всех проявлений мукополисахаридоза не неврологического характера.
Источник
Мукополисахаридоз, тип I (Синонимы: недостаточность фермента лизосомной a-L-идуронидазы, синдромы Гурлер, Гурлер-Шейе и Шейе).
Мукополисахаридоз, тип I – аутосомно-рецессивное заболевание, возникающее в результате снижения активности лизосомной a-L-идуронидазы, которая участвует в метаболизме гликозаминогликанов. Заболевание характеризуется прогрессирующими нарушениями со стороны внутренних органов, костной системы, психоневрологическими и сердечно-лёгочными расстройствами.
Код по МКБ-10
- Е76 Нарушения обмена гликозаминогликанов.
- Е76.0 Мукополисахаридоз, тип I.
Эпидемиология
Мукополисахаридоз I – панэтническое заболевание с частотой встречаемости в популяции в среднем 1 на 90 000 живых новорождённых. Средняя частота синдрома Гурлер в Канаде 1 на 100 000 живых новорождённых, синдрома Гурлер-Шейе – 1 на 115 000, синдрома Шейе – 1 на 500 000.
Классификация
В зависимости от выраженности клинических симптомов заболевания различают три формы мукополисахаридоза I: синдромы Гурлер, Гурлер-Шейе и Шейе.
Причины мукополисахаридоза I типа
Мукополисахаридоз I – аутосомно-рецессивное заболевание, возникающее в результате мутаций в структурном гене лизосомной альфа-L-идуронидазы.
Ген альфа-L-идуронидазы – IDUA – расположен на коротком плече хромосомы 4 в локусе 4р16.3. К настоящему времени известно более 100 различных мутаций в гене IDUA. Превалирующее число известных мутаций – точечные в различных экзонах гена IDUA. Для европеоидов характерны две частые мутации Q70X и W402X.
Самая распространённая мутация среди пациентов из российской популяции – мутация Q70X. Её частота – 57%, что сравнимо с частотой Q70X в скандинавской популяции (62%). Частота мутации W402X, которая встречается в 48% случаев мукополисахаридоза I в ряде европейских популяций, в российской популяции составляет 5,3%.
Патогенез мукополисахаридоза I типа
Фермент a-L-идуронидаза участвует в метаболизме двух гликозаминогликанов – дерматан-сульфата и гепарансульфата. Поскольку идуроновая кислота входит в состав дерматансульфата и гепарансульфата, при данном заболевании нарушен внутрилизосомный распад именно этих гликозаминогликанов, которые и накапливаются в лизосомах повсеместно: в хрящах, сухожилиях, надкостнице, эндокарде и сосудистой стенке, печени, селезёнке и нервной ткани. Отёк мягкой мозговой оболочки вызывает частичную окклюзию субарахноидальных пространств, что приводит к прогрессирующей внутренней и наружной гидроцефалии.
Поражаются клетки коры большого мозга, таламуса, ствола, передних рогов. Тугоподвижность суставов – результат деформации метафизов, утолщение суставной капсулы вторично по отношению к отложению в ней гликозаминогликанов и фиброзу. Обструкция дыхательных путей – следствие сужения трахеи, утолщения голосовых связок, избыточности отёчных тканей в верхних дыхательных путях.
Симптомы мукополисахаридоза I типа
Мукополисахаридоз, тип IH (синдром Гурлер)
У больных с синдромом Гурлер первые клинические признаки заболевания появляются на первом году жизни, с пиком манифестации от 6 до 12 мес. В ряде случаев уже с рождения наблюдают незначительное увеличение печени, пупочные или пахово-мошоночные грыжи. Обычно диагноз устанавливают в возрасте от 6 до 24 мес. Характерные изменения черт лица по типу гаргоилизма становятся очевидными к концу первого года жизни: большая голова, выступающие лобные бугры, широкая переносица, короткие носовые ходы с вывернутыми наружу ноздрями, полуоткрытый рот, большой язык, толстые губы, гиперплазия дёсен, нерегулярные зубы. Другие частые манифестные симптомы – тугоподвижность мелких и крупных суставов, кифоз поясничного отдела позвоночника (поясничный гибус), хронические отиты и частые инфекционные заболевания верхних дыхательных путей. Практически у всех пациентов с синдромом Гурлер, так же как и при других типах мукополисахаридоза, кожные покровы плотные на ощупь. Часто встречается гипертрихоз. У единичных больных в возрасте до 1 года заболевание дебютировало с развития острой сердечной недостаточности, вызванной эндокардиальным фиброэластозом. По мере прогрессирования заболевания присоединяются симптомы, свидетельствующие о вовлечении в патологический процесс внутренних органов, сердечно-лёгочной, центральной и периферической нервной систем. Ведущие неврологические симптомы – снижение интеллекта, задержка речевого развития, изменения мышечного тонуса, сухожильных рефлексов, поражение черепных нервов, комбинированная кондуктивная и нейросенсорная тугоухость. Прогрессирующая вентрикуломегалия часто приводит к развитию сообщающейся гидроцефалии. К концу первого и в начале второго года жизни появляются шумы в сердце, позднее формируются приобретённые аортальные и митральные пороки сердца. К концу второго года жизни выявляют гепатоспленомегалию и характерные скелетные нарушения по типу множественного дизостоза: короткую шею, задержку роста, тотальную платиспондилию, поясничный гибус, тугоподвижность мелких и крупных суставов, дисплазию тазобедренных суставов, вальгусную деформацию суставов, изменение со стороны кистей по типу «когтистой лапы», деформацию грудной клетки в виде бочкообразной или колоколообразной. Часто наблюдается прогрессирующее помутнение роговицы, мегалокорнеа, глаукома, застойные диски зрительных нервов и/ или их частичная атрофия.
Ранние рентгенологические признаки – деформация рёбер (по типу «вёсельных») и овоидная деформация тел позвонков, излишняя трабекуляция диафизов длинных трубчатых костей в сочетании с её недостаточностью в области метафизов и эпифизов. По мере прогрессирования заболевания формируется макроцефалия с утолщением костей свода черепа, преждевременным закрытием лямбдовидного и сагиттального швов черепа, уменьшение орбит, расширение спинки турецкого седла. Больные погибают обычно в возрасте до 10 лет от обструкции дыхательных путей, респираторных инфекций, сердечной недостаточности.
Мукополисахаридоз, тип I-H/S (синдром Гурлер-Шейе) Клинический фенотип синдрома Гурлер-Шейе занимает промежуточное положение между синдромами Гурлер и Шейе, для него характерны медленно прогрессирующие нарушения со стороны внутренних органов, костной системы, лёгкое снижение интеллекта или отсутствие такового. Заболевание обычно дебютирует в возрасте 2-4 лет. Основные клинические нарушения – поражение сердца и развитие обструктивного синдрома верхних дыхательных путей. У некоторых пациентов наблюдают тотальный спондилолистез, что может приводить к компрессии спинного мозга. Большинство пациентов доживают до третьего десятилетия жизни. Основная причина летального исхода – острая сердечно-сосудистая и лёгочная недостаточность.
Мукополисахаридоз, тип IS (синдром Шейе)
В первоначальной классификации мукополисахаридозов, до открытия первичного биохимического дефекта при синдроме Шейе, его выделяли в отдельный тип – мукополисахаридоз V. Синдром Шейе – наиболее мягкий по течению заболевания среди других форм мукополисахаридоза I, для него характерны тугоподвижность суставов, аортальные пороки сердца, помутнение роговицы и признаки множественного костного дизостоза. Первые симптомы обычно появляются в возрасте от 5 до 15 лет. Ведущие клинические симптомы – скелетные нарушения в виде тугоподвижности суставов с развитием карпального туннельного синдрома. Офтальмологические расстройства включают помутнение роговицы, глаукому и пигментную дегенерацию сетчатки. Нейросенсорная тугоухость – позднее осложнение заболевания. Обструктивный синдром верхних дыхательных путей часто приводит к возникновению апноэ во время сна, что в некоторых случаях требует установления трахеостомы. Миелопатия шейного отдела спинного мозга встречается реже, чем при синдроме Гурлер-Шейе. Часто отмечают стеноз аорты с недостаточностью кровообращения и гепатоспленомегалию. Интеллект при данном синдроме не страдает или наблюдают лёгкие когнитивные нарушения.
Диагностика мукополисахаридоза I типа
Лабораторные исследования
Подтверждающая биохимическая диагностика мукополисахаридоза I заключается в определении уровня экскреции гликозаминогликанов мочи и измерении активности лизосомной a-L-идуронидазы. Суммарная экскреция Ггликозаминогликанов в моче возрастает. Также наблюдают гиперэкскрецию дерматансульфата и гепарансульфата. Активность a-L-идуронидазы измеряется в лейкоцитах или культуре кожных фибробластов с использованием искусственного флюорогенного или хромогенного субстратов.
Пренатальная диагностика возможна путём измерения активности a-L-идуронидазы в биоптате ворсин хориона на 9-11-й неделе беременности и/или определения спектра ГАГ в амниотической жидкости на 20-22-й неделе беременности. Для семей с известным генотипом возможно проведение ДНК-диагностики.
Функциональные исследования
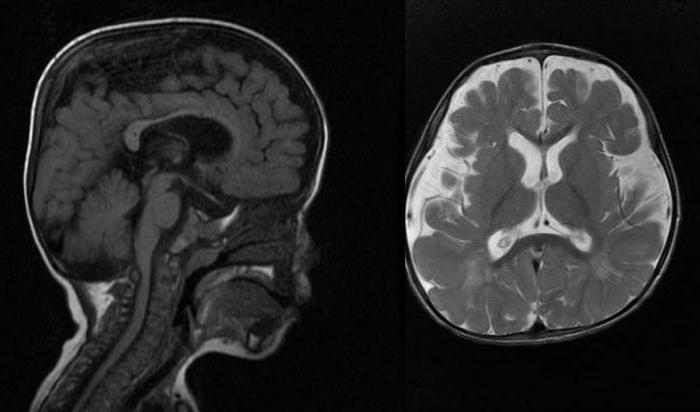
При рентгенографии у пациентов с синдромом Гурлер обнаруживают типичные признаки так называемого множественного костного дизостоза. При МРТ головного мозга обнаруживают множественные кисты в перивентрикулярных областях белого вещества головного мозга, мозолистого тела, реже базальных ганглиев, признаки гидроцефалии; в редких случаях – пороки головного мозга в виде лиссэнцефалии, мальформации Денди-Уокера.
Дифференциальная диагностика
Дифференциальная диагностика проводится как внутри группы мукополисахаридозов, так и с другими лизосомными болезнями накопления: муколипи-дозами, галактосиалидозом, сиалидозом, маннозидозом, фукозидозом, GM1-ганглиозидозом.
Лечение мукополисахаридоза I типа
При синдроме Гурлер показана трансплантация костного мозга, которая может кардинально изменить течение заболевания и улучшить его прогноз, однако эта процедура имеет много осложнений и проводится на ранних стадиях заболевания, преимущественно в возрасте до 1,5 лет. В настоящее время создан препарат для ферментозаместительной терапии мукополисахаридоза I – альдуразим (Aldurazyme, Genzyme), который зарегистрирован в странах Европы, США, Японии; его применяют для лечения экстраневральных нарушений при мукополисахаридозе I. Препарат показан для коррекции мягких форм мукополисахаридоза I (синдромах Гурлер-Шейе и Шейе). Препарат вводят еженедельно, внутривенно, капельно, медленно, в дозе 100 ЕД/кг. Для лечения синдрома Гурлер с выраженными неврологическими осложнениями препарат менее эффективен, поскольку фермент не проникает через гематоэнцефалический барьер.
[1], [2]
Источник