
Наследственная атрофия зрительного нерва синдром

Резкое двустороннее снижение остроты зрения может говорить о развитии опасной патологии — оптической нейропатии Лебера. Такое заболевание чаще затрагивает молодых людей мужского пола. Рассмотрим причины патологии, симптомы и особенности терапии в статье.
Впервые случаи заболевания, при котором острота зрения стремительно снижается из-за атрофии зрительного нерва, были описаны в 1871 году Теодором Лебером. Ученый наблюдал за развитием патологии у молодых людей из нескольких родственных семей, что указывало на наследственную причину заболевания. Механизм наследования болезни был тщательно изучен, поэтому удалось доказать, что патология передается по материнской линии преимущественно мужчинам.

До сих пор характер заболевания недостаточно хорошо изучен для того, чтобы удалось найти эффективные методы лечения такой болезни. Сейчас лечение больных заключается в поддерживающей терапии, но периодически группы ученых представляют новые методики генной терапии для клинических исследований, что, конечно, вселяет надежду людям с подобным диагнозом.
Оптическая нейропатия Лебера: симптомы, лечение болезни
Любые нарушения зрения, стремительно прогрессирующие, должны быть тщательно изучены и комплексно продиагностированы. Оптическая нейропатия Лебера редко сопровождается недомоганием, характерным для неврологических заболеваний, при этом зрение падает быстро, иногда даже не в течение нескольких месяцев, а за две-три недели. При такой остроте, зрение невозможно проверить по таблице Сивцева, потому что человек попросту не видит знаков, поэтому применяют аппаратную диагностику и метод «счета пальцев» на определенном расстоянии от лица пациента. Особенность заболевания в том, что зрительный нерв поражается из-за мутации гена. Вырабатывается избыточное количество токсичных молекул кислорода, что негативно сказывается на состоянии клеток нерва и зрение снижается.

Оптическая нейропатия Лебера — симптомы и особенности болезни:
молодой возраст пациентов — 18-35 лет;
болезнь чаще развивается у мужчин;
быстрая односторонняя, а затем и двусторонняя потеря зрения;
острота зрения снижается в течение нескольких недель;
нарушение цветового восприятия красного и зеленого цветов;
патология может сопровождаться симптомами, присущими митохондриальным болезням (судороги, нарушение проводимости сердца).
При атрофии зрительного нерва Лебера чаще всего процесс развития болезни начинается с одного глаза, но нередки случаи, когда поражаются сразу два глаза. Центральное зрение снижается постоянно. Это связано с постепенной смертью клеток зрительного нерва, передающего информацию об изображении в головной мозг. Иногда болезнь может сопровождаться симптомами, характерными для неврологических заболеваний. В этом случае больной человек страдает от дистонии, тремора, атаксии. Реже в начале развития патологии человек замечает искры, пятна, яркие вспышки перед глазами.
Причины наследственной оптической нейропатии Лебера
Причины нейропатии Лебера, при которой атрофируется зрительный нерв, — генетические. Болезнь, обусловленная специфической мутацией генов митохондрий, передается женщинами своему потомству. Мужчины и женщины наследуют патологию от матери. Почему так происходит? Огромное количество ДНК находится в ядре клеток и лишь небольшая его часть — в митохондриях. Гены ядра наследуются и от матери, и от отца, а вот гены митохондрии — только от матери. Мужчина, унаследовавший мутацию, не передаст ее своим потомкам.

У значительной части людей, которые являются носителем мутированного гена, болезнь не станет явной. Более 85% женщин и 50% мужчин, которые являются носителем гена синдрома LHON, болезнь не затронет. Причины, влияющие на прогрессирование заболевания, не ясны, но достоверно известно, что спровоцировать болезнь может неблагоприятная экологическая обстановка, стрессы, инфекции, токсическое воздействие табака и алкоголя.
Диагностика оптической нейропатии Лебера
Диагностика болезни осложнена схожестью ее симптомов с ишемической невропатией или с определенными патологиями — например, с рассеянным склерозом. Сложно отследить и анамнез, так как не у всех носителей мутировавшего гена развивается нейропатия Лебера. Расширенное офтальмологическое обследование может только установить наличие патологии, а вот для определения причины, понадобится генетическая диагностика.
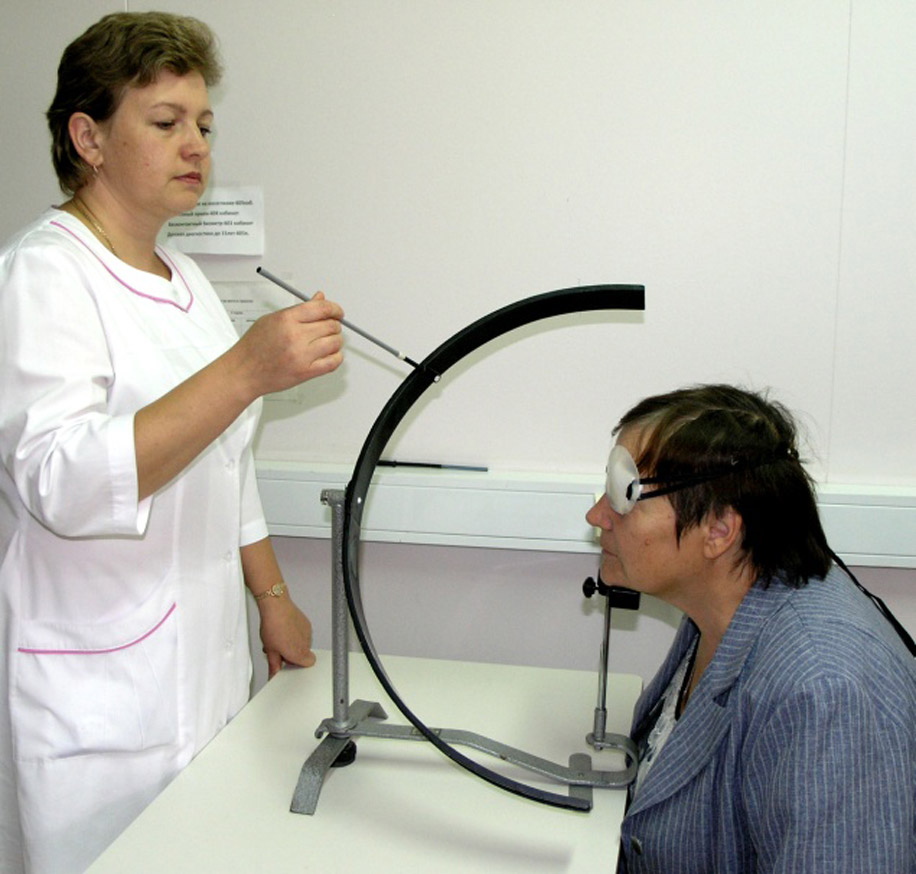
Особенности диагностики наследственной оптической нейропатии Лебера:
общее клиническое обследование;
анализ митохондриальной ДНК;
исследование полей зрения;
осмотр глазного дна;
когерентная томография и электроретинография (необходимы, чтобы исключить патологии сетчатки).
Специалистам рекомендуется проводить дифференциальную диагностику синдрома LHON, сравнивая симптоматику и данные обследования с другими заболеваниями, поражающими глазной нерв. Например, с ишемической, токсической невропатией. Полное офтальмологическое обследование показывает, что диск зрительного нерва воспаленный, также заметны телеангиэктазии сосудов (расширение мелких капилляров).
Наследственная оптическая нейропатия Лебера — можно ли вылечить болезнь?
Пока наследственная нейропатия оптическая считается неизлечимым заболеванием, но учеными ведется постоянный поиск новых путей для терапии подобной патологии. Благодаря методам генной терапии можно существенно замедлить прогрессирование болезней и улучшить качество жизни пациентов.
Стремительное прогрессирование болезни Лебера приводит к быстрой потере зрения людьми, которые всю свою жизнь видели нормально и не имели проблем со зрением. Неутешительный диагноз, а также информация о том, что болезнь пока не лечится, серьезно сказываются на самочувствии таких пациентов, которые не были готовы к инвалидности. Многие из существующих методик лечения патологии оказались малоэффективными, в том числе и хирургическое вмешательство.
К счастью, ученые, занимающиеся редкими патологиями, прикладывают немало усилий к тому, чтобы найти средство излечения. Особую надежду специалисты возлагают на генную терапию, и у многих исследовательских групп уже есть неплохие результаты. Иногда препятствием к продолжению экспериментов является их этическая сторона, так как дальнейшие исследования предполагают использование разработанной методики на людях. Есть ли успехи хотя бы с использованием экспериментальных моделей? Да, генетикам уже удалось найти путь к решению проблемы.
Так, группа ученых из Майами, используя экспериментальные модели, доказала, что мутировавшие гены можно безопасно заменить здоровыми, это предотвратит ухудшение питания клеток зрительного нерва. Для исправления генетического дефекта в митохондрии необходимо ввести нормальную ДНК — это позволит исправить нарушение и восстановить зрительную функцию. Ученые сообщают, что такой подход будет эффективным и в отношении других заболеваний, вызванных митохондриальными мутациями, а также различных нарушений, связанных с процессами старения организма.
Нейропатия Лебера: лечение и поддерживающая терапия
На данный момент медики могут предложить поддерживающую терапию, которая замедляет прогрессирование патологии. Пациентам необходимо кардинально пересмотреть уклад жизни, снизить уровень стресса и влияние негативных факторов на здоровье, отказаться от курения и алкоголя.
О появившейся проблеме обычно сигнализирует отек слоя нервных волокон (диагностика с использованием когерентной томографии). Также обнаружить изменения помогут цветовые тесты, которые сообщают о проблеме с распознаванием зеленого и красного цветов. Людям, у которых в семье были случаи оптической нейропатии Лебера, необходимо не забывать о высоких рисках развития этой опасной патологии.
Существуют разные методы терапии заболевания и пока ни один из них не имеет доказанной эффективности. Лечение ведется специалистами с учетом степени нарушения, а также имеющихся сопутствующих заболеваний у пациента, поэтому используются комбинированные протоколы терапии.

Поддерживающая терапия при атрофии зрительного нерва:
стимуляция кровообращения в сохранившихся нервных волокнах;
применение сосудорасширяющих средств, стимуляторов;
лечение нейропротекторами (средствами, предупреждающими повреждение нейронов мозга);
применение препаратов, улучшающих обменные процессы на клеточном уровне;
физиотерапевтические процедуры, ультразвуковая терапия, электрофорез с лекарственными препаратами и травами, магнитотерапия, электростимуляция зрительного нерва;
сбалансированное питание — в рацион необходимо включить продукты с высоким содержанием витаминов С, B1, B12;
биостимуляторы (алоэ, экстракт стекловидного тела);
комплексные витаминные препараты, глазные капли с витаминами.
Что касается прогноза заболевания, то в каждом случае он индивидуален, зачастую все зависит от возраста пациента. Чем моложе заболевший человек, тем выше шансы на то, что зрение хотя бы частично восстановится. Полная слепота развивается в редких случаях. При таком заболевании важно соблюдать все предписания специалиста, ограничить воздействие токсинов на организм (отказаться от вредных привычек, исключить прием токсичных лекарственных препаратов, некоторых видов антибиотиков).
Сложности в поддерживающей терапией возникают обычно на местах, ведь многие врачи не учитывают генетическую причину болезни как основную и ставят ошибочные диагнозы. Повышение уровня информированности о таком заболевании, как оптическая нейропатия Лебера, значительно увеличивает шансы на то, что на эту проблему будет обращено внимание разных специалистов и ученых генетиков.
Источник
Наследственные атрофии зрительного нерва при неврологических и системных болезняха) Аутосомно-доминантная атрофия зрительного нерва и сенсоневральная тугоухость. Описано несколько семей с аутосомно-доминантной атрофией зрительного нерва, сочетающейся с глухотой. Во многих из этих семей не наблюдается других системных или неврологических аномалий. В некоторых, но не во всех этих семьях было выявлено носительство мутаций гена ОРА1, которые в других семьях вызывают изолированную нейрооптикопатию. В одной итальянской семье был идентифицирован новый локус хромосомы 16 (16q21-q22), обозначенный ОРА8, предварительные исследования указывают на то, что в патогенезе также может играть роль митохондриальная дисфункция. В голландской семье, наследующей доминантную атрофию зрительного нерва и глухоту, мутации ОРА1 были исключены, но была выявлена ранее неизвестная миссенс-мутация гена WFS1 хромосомы 4 (4р16.1), в локусе, мутации которого обычно вызывают аутосомно-рецессивный DIDMOAD-синдром, он же синдром Wolfram, характеризующийся синдромальной нейрооптикопатией, сахарным диабетом, несахарным диабетом и тугоухостью. Аналогично в другой семье, наследующей доминантную атрофию зрительного нерва с тугоухостью и нарушением регуляции глюкозы, при анализе мутаций были исключены мутации генов ОРА1, ОРАЗ, ОРА4 и ОРА5, но была выявлена новая миссенс-мутация гена WFS1 (синдрома Wolfram). В других семьях доминантной атрофии зрительного нерва и глухоте сопутствовали атаксия, слабость конечностей или полинейропатия. Может встречаться тяжелое ухудшение слуха при рождении и задержка речевого развития, или же умеренная медленно прогрессирующая тугоухость. Была предложена аббревиатура CAPOS (cerebellar ataxia, areflexia, pes cavus, optic atrophy, sensorineural deafness — мозжечковая атаксия, арефлексия, полая стопа, сенсоневральная тугоухость), но данная нозология все еще генетически не определена. Возможно, во многих из этих семей наследуемые сложные синдромы являются формами «доминантной атрофии зрительного нерва плюс», вызванной мутациями OРА1, но ясно, что синдромальное сочетание доминантной атрофии зрительного нерва и тугоухости является генетически гетерогенной группой состояний. б) Аутосомно-доминантная атрофия зрительного нерва, глухота, офтальмоплегия и миопатия. Сочетание аутосомно-доминантной атрофии зрительного нерва, глухоты, птоза, офтальмоплегии, дистаксии и миопатии должно вызывать подозрение о наличии «доминантной атрофии зрительного нерва плюс» и является показанием для анализа на мутации гена ОРА1 хромосомы 3. Ухудшение зрения обычно начинается в первом десятилетии жизни и прогрессирует до значений от 20/30 до 20/400, главным образом вследствие нейрооптикопатии, но также могут выявляться аномалии на электроретинограмме. Сенсоневральная тугоухость манифестирует в первом или втором десятилетии жизни и прогрессирует. Офтальмоплегия и миопатия развиваются в среднем возрасте. Эта патология представляет собой митохондриальное заболевание, развивающееся вследствие ядерных генетических аномалий. в) Аутосомно-доминантная атрофия зрительного нерва с рано развивающейся катарактой. В двух семьях из Франции наблюдалось наследуемое по аутосомно-доминантному типу сочетание атрофии зрительного нерва и рано развивающейся катаракты. Мутации гена ОРА1 были исключены, но были выявлены патогенные мутации гена ОРАЗ хромосомы 19 (19q13.2-q13.3), локус этих мутаций обычно вызывает синдром Costeff, синдромальную нейрооптикопатию, наследуемую по аутосомно-рецессивному типу. При скрининговых исследованиях на мутации ОРА3 по поводу моносимптомных доминантных атрофий зрительного нерва в различных семьях не удалось выявить какие-либо патогенные варианты ОРАЗ; вероятно, мутации ОРАЗ, вызывающие доминантную атрофию зрительного нерва, встречаются крайне редко. г) Аутосомно-рецессивная атрофия зрительного нерва в сочетании с прогрессирующей нейродегенерацией и 3-метилглютакониковой ацидурией III типа (синдром Costeff). При этом аутосомно-рецессивном синдроме, чаще всего встречающемся в еврейских семьях Ирака, тяжелая атрофия зрительного нерва сопутствует экстрапирамидным расстройствам, когнитивным нарушениям, повышению в моче уровня 3-метилглютакониковой кислоты и повышению в плазме уровней 3-метилглютаровой кислоты. Вызывающий заболевание ген был локализован на хромосоме 19 (19q13.2—q13.3) и получил название ОРА3.
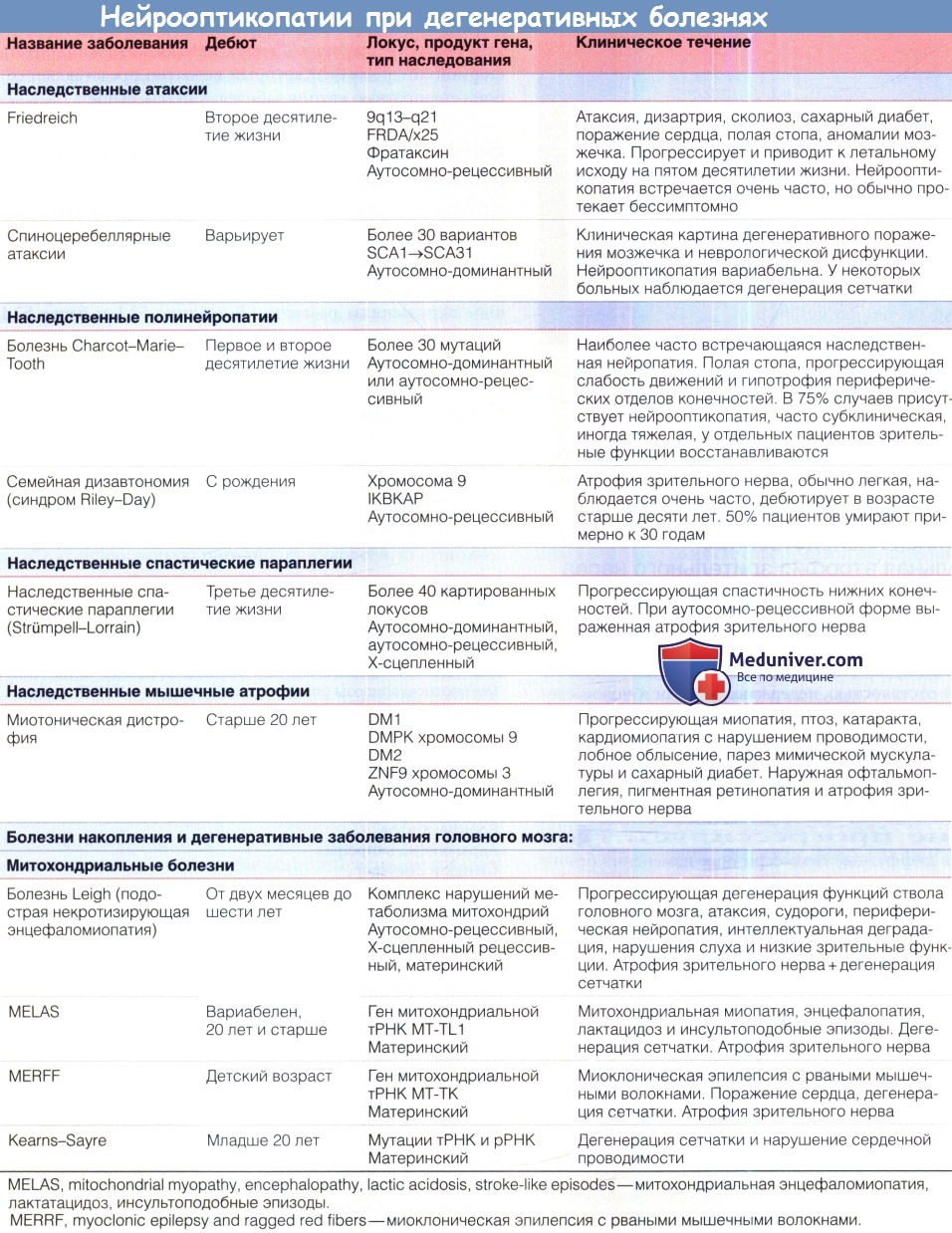
д) Аутосомно-рецессивная атрофия зрительного нерва в сочетании с ювенильным сахарным диабетом, несахарным диабетом и тугоухостью (синдром Wolfram). Этот синдром включает в себя ювенильный сахарный диабет и прогрессирующее ухудшение зрения на фоне атрофии зрительного нерва, почти всегда сопровождающихся несахарным диабетом, нейросенсорной тугоухостью или и тем, и другим (вследствие чего был предложен эпоним DIDMOAD — diabetes insipidus, diabetes mellitus, optic atrophy, deafness, т. e. несахарный диабет, сахарный диабет, атрофия зрительного нерва, глухота). Сахарный диабет развивается в течение первого или второго десятилетия жизни и обычно предшествует развитию атрофии зрительного нерва. Однако у нескольких пациентов атрофия зрительного нерва и ухудшение зрения являлись первыми проявлениями синдрома. На ранних стадиях заболевания острота зрения может оставаться нормальной, несмотря на легкую дисхроматопсию и атрофию зрительного нерва. На поздних стадиях развивается тяжелое ухудшение зрения. При периметрии выявляются генерализованное сужение и центральные скотомы поля зрения. Всегда развивается тяжелая атрофия зрительного нерва, может наблюдаться маленькая или средних размеров экскавация ДЗН. И ухудшение слуха, и несахарный диабет дебютируют в первом или во втором десятилетии жизни и могут протекать в очень тяжелой форме. У половины пациентов присутствует атония мочевыводящих путей, сопровождающаяся рецидивирующими инфекциями мочевых путей, нейрогенным недержанием и даже смертельными осложнениями. Другие системные и неврологические нарушения включают в себя атаксию, осевую ригидность, судороги, стартл-миоклонус, тремор, нарушение моторики желудочно-кишечного тракта, нарушение вестибулярных функций, центральное апноэ, нейрогенный коллапс верхних дыхательных путей, птоз, катаракту, пигментную ретинопатию, ирит, снижение слезопродукции, зрачки Adie, офтальмоплегию, недостаточность конвергенции, паралич вертикального взора, нистагм, умственную отсталость, психиатрические нарушения, маленький рост, первичную гонадную атрофию, другие эндокринные аномалии, аносмию, мегалобластную и сидеробластную анемию, аномалии при электроретинографии и повышение содержания белка в цереброспинальной жидкости. При лучевых исследованиях и на вскрытии у некоторых пациентов выявляются обширные атрофические изменения и мальформации развития коры, что указывает на диффузное нейродегенеративное расстройство с преимущественным поражением среднего мозга и моста. Если синдром сопровождается анемией, терапия тиамином может облегчить анемию и снизить потребность в инсулине. В нескольких семьях методом анализа сцепления удалось локализовать ген синдрома Wolfram на хромосоме 4 (4р16.1), который был обозначен как WFS1; были описаны точечные мутации и делеции этого гена. Продукт данного гена — вольфрамин — белок эндоплазматического ретикулума, участвующий в регуляции уровня внутриклеточного кальция. Идентифицирован второй вызывающий синдром Wolfram ген, локализованный на другом плече хромосомы 4 (4q22-24), он получил обозначение CISD2. У больных наблюдается повышенная кровоточивость и пептические язвы. Нокаут гена CISD2 у мышей вызывает развитие Wolfram-подобного синдрома, сопровождающегося преждевременным старением вследствие поражения митохондрий. В общем, многие из сопутствующих аномалий, описанных при синдроме Wolfram, обычно встречаются у пациентов с предположительно митохондриальной патологией, особенно у больных с синдромами хронической прогрессирующей наружной офтальмоплегии. На основании этих данных было высказано предположение, что фенотип Wolfram, возможно, представляет собой не специфическую аномалию, а является проявлением различных патогенных ядерных или митохондриальных генетических дефектов, в конечном итоге запускающих общий механизм митохондриальной дисфункции. К тому же большинство случаев синдрома Wolfram классифицировались как спорадические или наследуемые по рецессивному типу, последний вывод делался на основании экспрессии синдрома у сиблингов (что, как сейчас известно, также соответствует и материнскому типу наследования). е) Спастическая параплегия, атрофия зрительного нерва и нейропатия (синдром SPOAN — spastic paraplegia, optic atrophy, neuropathy). Аутосомно-рецессивное нейродегенеративное заболевание было описано клинически как сочетание непрогрессирующей врожденной атрофии зрительного нерва, дебютирующей в младенческом возрасте спастической параплегии, дебютирующего в детстве прогрессирующего поражения двигательных и чувствительных аксонов, дебютирующей в третьем десятилетии жизни дизартрией, выраженных слуховых стартл-реакций, прогрессирующих контрактур суставов и деформации позвоночника. Выявлено сцепление с хромосомой 11q13, но патогенный ген все еще не установлен. ж) Врожденная мозжечковая атаксия, умственная отсталость, атрофия зрительного нерва и аномалии кожи (congenital cerebellar ataxia, mental retardation, optic atrophy, skin abnormalities— CAMOS). Непрогрессирующая аутосомно-рецессивная врожденная атаксия в сочетании с атрофией зрительного нерва, тяжелой умственной отсталостью и аномалиями строения кожи; заболевание сцеплено с локусом хромосомы 15 (15q24-q26), но патогенный ген все еще не выявлен. з) Глухота, дистония и нейрооптикопатия (deafness, dystonia, optic neuropathy—DDON, синдром Mohr-Tranebjaerg). Это Х-сцепленное заболевание манифестирует в старшем детском возрасте сенсоневральной тугоухостью, дистонией и атаксией, после чего в возрасте около двадцати лет наблюдается атрофия зрительного нерва, а до пятидесяти лет развиваются снижение интеллекта и психиатрические нарушения. Прогноз для зрения неблагоприятный, большинство пациентов слепнут в возрасте около сорока лет. Причиной заболевания являются мутации гена TIMM8АХ-хромосомы (Xq22), продукт гена локализуется в межмембранном пространстве митохондрий. Было выявлено нарушение биохимических процессов в митохондриях. и) Осложненная наследственная инфантильная атрофия зрительного нерва (синдром Behr). Описание синдрома Behr включает в себя манифестирующую в детстве атрофию зрительного нерва, в сочетании с различными нарушениями пирамидного тракта, атаксией, умственной отсталостью, недержанием мочи и полой стопой. Синдром обычно наследуется по аутосомно-рецессивному типу, поражаются представители обоего пола. Ухудшение зрения, от умеренного до тяжелого, обычно развивается в возрасте младше десяти лет, часто сопровождается нистагмом. В большинстве случаев по прошествии периода детства патология не прогрессирует. При лучевых исследованиях выявляются диффузные симметричные изменения белого вещества. У некоторых пациентов с синдромом Behr клиническая картина может напоминать проявления наследственной атаксии. Вероятно, синдром Behr является гетерогенным заболеванием, развивающимся вследствие различных этиологических, в том числе генетических, факторов. к) Прогрессирующая энцефалопатия с отеком, гипсаритмия и атрофия зрительного нерва (синдром РЕНО — progressive encephalopathy with edema, hypsarrhythmia, optic atrophy). Описаны прогрессирующая энцефалопатия, манифестирующая в первые полгода жизни, вслед за которой развиваются тяжелая гипотония, судороги с гипсаритмией, тяжелая умственная отсталость, гиперрефлексия, транзиторный или персистирующий отек лица и тела, атрофия зрительного нерва. Последняя обычно выявляется на первом или втором году жизни, часто наблюдается нистагм. Дефект метаболизма еще не установлен; вероятно, заболевание наследуется по аутосо-мно-рецессивному типу. Данную патологию можно считать формой синдрома Behr, который, вероятно, представляет собой гетерогенную группу заболеваний. л) Нейрооптикопатия как проявление наследственных дегенеративных заболеваний или патологии развития. Нейрооптикопатия может сопутствовать различным наследственным дегенеративным заболеваниям или системным нарушениям развития. В таблице ниже перечислены основные проявления наиболее часто встречающихся из них.
– Также рекомендуем “Неврит зрительного нерва у ребенка” Оглавление темы “Патология зрительного нерва.”:
|


Источник