
Нерасхождение хромосом человека и их синдромы

Хромосомные аномалии. Аномалии количества и качества
Хромосомные аномалии подразделяют на две категории — количественные и структурные. При количественных аномалиях общее количество хромосом отличается от нормального (46 хромосом). При структурных аномалиях происходит физическая перестройка хромосом.
Хромосомы состоят из коротких плеч (р) и длинных плеч (q). У них есть первичная перетяжка, или центромера, к которой прикрепляются микротрубочки при делении клетки. Теломеры, или верхушки хромосом, покрыты повторяющейся последовательностью TTAGGG, крайне важной для поддержания целостности хромосомы. Положение центромеры определяет строение хромосомы. У человека акроцентрические хромосомы 13, 14, 15, 21 и 22 характеризуются наличием стебельков и спутников, где расположены гены рибосомной РНК.
Аномалии количества хромосом. Аномалии количества хромосом — наиболее хорошо диагностируемые из хромосомных аномалий. Структурные аномалии играют большую роль в развитии врожденных дефектов, бесплодия и привычных выкидышей. Количественные аномалии хромосом, как правило, возникают из-за ошибок при делении клеток, когда происходит увеличение и/или уменьшение хромосом в дочерних клетках, или нерасхождение хромосом. Нерасхождение может возникать как в митозе, так и в мейозе. Клетки, получающиеся в результате такого деления, бывают анеуплоидными, так как их хромосомный набор не соответствует типичному гаплоидному набору из 23 хромосом.
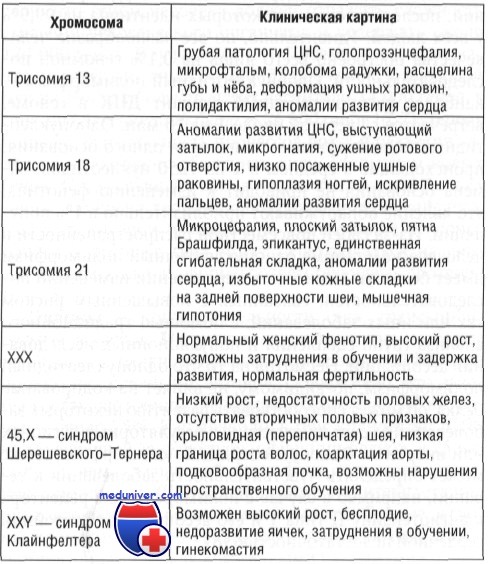
Нерасхождение может происходить как в первом, так и во втором делении мейоза. И в том, и в другом случае возможно анеуплоидное оплодотворение. Нерасхождение или запаздывание хромосом в анафазе может случиться в митозе, что вызывает мозаицизм, т.е. наличие двух и более клеточных линий у одного индивида. Часто мозаицизм обнаруживают при аномалии половых хромосом — синдроме Шерешевского-Тернера (до 50% случаев). Наиболее распространенные количественные хромосомные аномалии перечислены в табл. 5-1. Количественные аномалии также могут характеризоваться кратным гаплоидному набору (23) числом хромосом.
Триплоидия (с количеством хромосом 69) обычно возникает при оплодотворении одной яйцеклетки сразу двумя сперматозоидами. Триплоидные оплодотворения обнаруживают в 15% случаев спонтанных выкидышей, связанных с хромосомными аномалиями; лишь иногда они завершаются родами. Тетраплоидия характеризуется наличием 92 хромосом в наборе и встречается в небольшом проценте спонтанных абортов.
Аномалии структуры хромосом. Структурные перестройки хромосом происходят при таких хромосомных повреждениях, при которых по каким-либо причинам не происходит восстановления их изначальной структуры. Хромосомные перестройки бывают сбалансированными, если сохраняется диплоидный генетический набор клетки. Несбалансированные перестройки вызывают анеуплоидию в одном сегменте хромосом или более. Такие структурные перестройки могут накапливаться (тогда они называются семейными) или возникать как первичное событие, de novo.
Сбалансированные семейные хромосомные перестройки в большинстве случаев бывают истинно сбалансированными и не представляют существенного риска развития врожденных дефектов или задержки умственного развития. Хромосомные перестройки de novo, которые выглядят сбалансированными, обусловливают небольшой риск анеуплоидии на молекулярном уровне (5%), вызывая врожденные дефекты и задержку развития.
Транслокация — обмен плечами между двумя разными хромосомами. Реципрокные транслокации происходят при разрывах, ограничивающихся двумя плечами, и реципрокном обмене дистальными сегментами, приводящем к образованию производной хромосомы. В большинстве случаев фенотип носителей сбалансированных транслокаций вполне нормален, но риск образования несбалансированных гамет при гаметогенезе у них повышен. У носителей сбалансированных транслокаций возможен сложный тип расщепления хромосом, включающий нормальный паттерн расщепления, сбалансированный транслокационный паттерн и несбалансированный паттерн, приводящий к частичной трисомии и частичной моносомии заинтересованных хромосом.
Когда в транслокации участвуют короткие плечи двух акроцентрических хромосом, длинные плечи соединяются в центромерном участке одной хромосомы, при этом короткие плечи акроцентрических хромосом утрачиваются. Это так называемые робертсоновские транслокации. Короткие плечи акроцентрических хромосом содержат резервный генетический материал рибосом, поэтому его утрата на фенотипе не отражается. Как и при сбалансированных транслокациях, продукты мейотического расхождения могут быть как сбалансированными, так и несбалансированными.
Другие структурные аномалии могут приводить к невынашиванию беременности и врожденным дефектам. Если на одной хромосоме образуется два разрыва, и в процессе репарации этот участок поворачивается на 180°, происходит инверсия. Если такой процесс случается в каждом плече хромосомы, это называется перицентрической инверсией. Если же в инверсию вовлекается только одно плечо, это парацентрическая инверсия. Каждая из этих разновидностей инверсий оказывает уникальное и различное влияние на гаметогенез и вынашивание беременности. При хромосомных дупликациях происходит удвоение сегмента хромосомы различной длины, что приводит к развитию частичной трисомии по этому сегменту. При делециях сегмент различной длины теряется, что приводит к генетическому дисбалансу – частичной моносомии.
Такое состояние, при котором второй экземпляр гена или сегмента хромосомы отсутствует, что выражается в патологическом фенотипе или клиническом синдроме, известно под названием гаплонедостаточности.
– Также рекомендуем “Флюоресцентная гибридизация in situ. Применение технологии FISH”
Оглавление темы “Сперматогенез”:
- Формирование сперматозоидов. Сперматоцитогенез и спермиогенез
- Цикл сперматогенеза. За какое время созревают сперматозоиды?
- Строение сперматозоидов. Структура
- Регуляция сперматогенеза. Факторы влияющие на формирование сперматозоидов
- Функции придатка яичка. Транспорт и хранение спермы
- Проникновение сперматозоидов через шейку матки. Капациитация и акросомальная реакция
- Геном человека. Один ген – один белок – справедливо ли?
- Гены и хромосомы человека. Строение
- Хромосомные аномалии. Аномалии количества и качества
- Флюоресцентная гибридизация in situ. Применение технологии FISH
Источник
Хромосомные аномалии включают аномалии количества (количественные аномалии) или структуры хромосом (структурные аномалии) и является важным фактором самопроизвольных выкидышей и врожденных аномалий развития. Приводятся данные, что 50% всех оплодотворений завершаются самопроизвольными выкидышами и в 50% случаев этих выкидышей имеющиеся хромосомные аномалии. Наиболее частыми хромосомными аномалиями у абортусов есть 45, Х (синдром Тернера, или Шерешевского – Тернера), триплоидия и трисомия 16. Хромосомные аномалии являются причиной 7% крупных врожденных пороков развития, а мутации генов — около 8%.
- Количественные аномалии хромосом
- Трисомия 21
- Трисомия 18
- Синдром Кляйнфельтера (ХХV или XXXV)
- Синдром Тернера, Шерешевского – Тернерa
- Структурные аномалии хромосом
- Методы генетического анализа
Количественные аномалии хромосом
Нормальная соматическая клетка является диплоидной и содержит 46 хромосом, или 2n хромосом. Нормальные гаметы является гаплоидными и содержат n хромосом. Еуплоидия означает любое целое число n, например, диплоидию или триплоидию. Анеуплоидия означает любое целое число хромосом, которое не является эуплоидным и чаще означает наличие лишней хромосомы (трисомия) или потерю одной хромосомы (моносомия).
Аномалии количества хромосом могут возникать во время мейотического или митотического делений. В ходе мейоза в норме обе хромосомы из гомологичной пары отделяются первое мейотическое деление и каждая дочерняя клетка получает один компонент из каждой пары хромосом. Если отделение гомологичных хромосом не происходит (нерасхождение хромосом), обе хромосомы с гомологичной пары попадают в одну клетку. Вследствие нерасхождения хромосом одна клетка получает 24 хромосомы, а вторая — 22 хромосомы.
Когда при оплодотворении гамета, содержащего 23 хромосомы, соединяется с гаметой, содержащей 24 или 22 хромосомы, образуется зигота с 47 (трисомия) или 45 (моносомия) хромосомами. Нерасхождение может происходить во время первого или второго мейотического деления и касаться как соматических (аутосом), так и половых хромосом. Частота хромосомных аномалий, в том числе нерасхождения хромосом, увеличивается в оогенезе у женщин старше 35 лет.
Нерасхождение хромосом во время митоза (митотическое нерасхождение) клеток эмбриона на самых ранних стадиях эмбриогенеза, когда одни клетки получают аномальное количество хромосом, другие — нормальное, получило название мозаицизма. Индивиды с мозаицизмом хромосом могут иметь лишь отдельные проявления того или иного синдрома зависимости от количества и распределения пораженных клеток.
Иногда возникает разрыв хромосомы, и части одной хромосомы присоединяются к другой — транслокация хромосом. Такие транслокации могут быть сбалансированными и несбалансированными. При сбалансированных транслокациях разрыв и объединения двух хромосом происходят без потери важного генетического материала, и индивид является нормальным. Несбалансированные транслокации характеризуются потерей части одной из хромосом и изменением генотипа. Так, например, несбалансированные транслокации между длинными плечами хромосом 14 и 21 во время мейоза I или II вызывают образование гамет с лишней 21-й хромосомой (трисомия 21), что является одной из причин возникновения синдрома Дауна.
Трисомия 21
Трисомия 21 (синдром Дауна) чаще всего обусловлена наличием лишней копии хромосомы 21 (трисомия 21). Детям с синдромом Дауна свойственны:
- задержка роста,
- различные степени умственного отставания,
- черепно-лицевые аномалии (эпикант, скошенный разрез глаз, плоское лицо, маленькие низко размещенные уши),
- пороки развития сердца,
- гипотония и др.
У таких детей чаще имеют место инфекции, дисфункции щитовидной железы, преждевременное старение, развитие болезни Альцгеймера в молодом возрасте (с 35 лет). В 95% случаев синдром обусловлен трисомией 21 через мейотическое неразличие, в 75% случаев нерасхождения случается во время оогенеза. Частота синдрома Дауна составляет 1: 2000 оплодотворений у женщин в возрасте <25 лет и возрастает до 1 300 – в возрасте 35 лет и 1 100 – в возрасте 40 лет.
Примерно в 4% случаев синдрома Дауна наблюдается несбалансированная транслокация между хромосомой 21 и одной из хромосом 13, 14 или 15. В 1% случаев синдром Дауна может быть обусловлен мозаицизмом в результате митотического нерасхождения. Часть клеток таких индивидуумов имеют нормальное количество хромосом, другие анэуплоидное. В таких индивидуумов может проявляться различное количество признаков синдрома Дауна в зависимости от количества и локализации аномальных клеток.
Трисомия 18
Трисомия 18 (синдром Эдвардса) встречается с частотой 1: 5000 новорожденных и характеризуется умственной отсталостью, микрогнатией, врожденными пороками сердца, почек, скелета, согнутыми пальцами и синдактилией. Такие дети обычно умирают в возрасте до 2 мес.
Трисомия 13 (синдром Патау) является редким заболеванием (1:15 500 живых новорожденных) и включает умственную отсталость, голопрозэнцефалию, врожденные пороки сердца, глухоту, расщелины губы и неба, глазные недостатки (микрофтальмия, анофтальмия, колобома). Большинство детей умирают в возрасте до 3 мес.
Синдром Кляйнфельтера (ХХ
V
или XXXV)
Проявляется у лиц мужского пола в период полового созревания и характеризуется атрофией яичек, гиалинизацией семенных канальцев, бесплодием и, часто, гинекомастией. Частота синдрома Кляйнфельтера составляет 1: 500 мужчин. Клетки содержат 47 хромосом с дополнительной Х-хромосомой (генотип ХХУ) вследствие нерасхождения ХХ гомологичных хромосом. Тельца полового хроматина оказываются в 80% случаев. Иногда больные с синдромом Кляйнфельтера имеют 48 хромосом, то есть 44 аутосомы и 4 половые хромосомы (ХХХУ). Синдром обычно не сопровождается задержкой умственного развития, но чем больше Х хромосом у генотипе, тем больше риск развития умственной отсталости.
Синдром Тернера, Шерешевского – Тернера (ХО, структурные аномалии или мозаицизм)
Синдром Тернера характеризуется женским генотипом при отсутствии яичников (дисгенезия гонад, или гонадный дисгенез). Больные имеют низкий рост, широкую грудную клетку с большим расстоянием между сосками, деформации скелета, перепончатую шею, лимфедему конечностей. Примерно в 55% случаев имеет место моносомия Х (ХО), половой хроматин в клетках отсутствует.
В 80% случаев причиной развития синдрома является дефект сперматозоида (нерасхождения половых хромосом в ходе сперматогенеза). В остальных случаях имеющиеся структурные аномалии Х-хромосомы или мозаицизм вследствие митотического нерасхождения.
Синдром трисомии Х (ХХХ) характеризуется определенной степенью умственной отсталости, задержкой полового развития, гипоменореей. В клетках содержатся по 2 тельца полового хроматина.
Структурные аномалии хромосом
Структурные аномалии хромосом могут касаться одной или более хромосом и обычно возникают через разрывы хромосом. Разрывы хромосом могут быть вызваны действием агрессивных факторов внешней среды: вирусов, радиации или химических веществ. Последствия разрыва хромосом зависят от дальнейшей судьбы разорванных частиц. В некоторых случаях оторвана часть хромосомы теряется — частичная делеция хромосомы. В случаях частичной делеции хромосомы ребенок характеризуется определенными аномалиями.
Синдром «кошачьего крика» обусловлен частичной делецией короткого плеча хромосомы 5 и характеризуется микроцефалией, умственной отсталостью, врожденными пороками сердца. Плач таких детей напоминает кошачий крик.
Микроделеции хромосом охватывают лишь несколько смежных генов и могут приводить к развитию синдрома микроделеции, или синдрома смежных генов. Места возникновения таких делеций называют смежными генными комплексами. Их идентифицируют с помощью высокотехнологичных дифференциальной окраски хромосомных полос.
Примером может быть микроделеция длинного плеча хромосомы 15 (15 ^ 11-15 ^ 13). При наследовании пораженной делецией материнской хромосомы возникает синдром Ангельмана, при котором ребенку присуща умственная отсталость, задержка речевого и моторного развития, непровоцированные приступы смеха. При наследовании дефектной родительской хромосомы возникает синдром Прадера-Вилли. Пораженные индивиды страдают от гипотонии, ожирения, умственной отсталости, гипогонадизма и крипторхизма. Такое различие экспрессии генетического материала в зависимости от наследования родительской или материнской хромосомы является примером геномного импринтинга.
Другими генными синдромами, которые могут наследоваться от обоих родителей, является синдром Миллера-Дикер в результате делеции 17р 13 (задержка развития, врожденные пороки лица и сердца) и синдром Шпринтцена при делеции 22 ^ 11 (дефекты неба, пороки развития сердца, задержка речевого развития, нарушение способности к обучению, шизофреноподобные расстройства).
Ломкие места — это участки хромосом, проявляющих склонность к отрыву или разрушения при определенных манипуляциях с клеткой. Ломкие места можно обнаружить при культивировании лимфоцитов в среде с дефицитом фолатов. Синдром ломкой Х-хромосомы сопровождается умственной отсталостью, большими ушами, выступание челюсти и бледными голубыми радужками.
Мужчины поражаются чаще, чем женщины (4/2000 сравнению с 1/2000), чем объясняется преобладание мужского пола среди умственно отсталых лиц. Синдром ломкой Х-хромосомы занимает второе место после синдрома Дауна среди причин умственной отсталости хромосомного происхождения.
Генные мутации
Многие врожденные пороки человека является унаследованными; некоторые из них наследуются строго по законам Менделя. Во многих случаях дефект является прямым следствием изменения структуры или функции единственного гена — мутации одного гена, моногенные мутации. Этот тип дефектов составляет около 8% случаев всех пороков человека.
За исключением Х и У-хромосом, у лиц мужского пола гены представлены парами, или аллелями, поэтому каждая генетическая детерминанта существует в двух нуклеотидных последовательностях, одна из которых происходит от отца, вторая — от матери. Если мутантный ген вызывает аномалию при повреждении только одной нуклеотидной последовательности, независимо от присутствия нормального аллеля, это получило название доминантного мутации. Если для появления недостатки оба аллеля должны быть аномальными (двойное повреждение), или если эта мутация происходит у мужчин и связана с Х-хромосомой, она называется рецессивной мутацией. Степени проявлений мутантных генов зависят от действия повреждающих факторов.
Использование методов молекулярной биологии позволило определить гены, ответственные за нормальное развитие. Картирование генома человека дает информацию о позиции многих из этих генов, а последующий анализ позволяет выявить их мутации. Эти данные позволяют расшифровать роль отдельных генов и их мутаций в развитии различных клинических синдромов и заболеваний.
Кроме возникновения врожденных пороков, дефектные гены обусловливают значительное количество врожденных нарушений метаболизма. Эти болезни (фенилкетонурия, гомоцистинурия и галактоземия) нередко сопровождаются различными степенями умственной отсталости или приводят к ним.
Методы генетического анализа
Идентификация генетических аномалий проводится с помощью метода полосок Гимза (С-полосок). Согласно этому методу, хромосомные препараты обрабатывают трипсином и окрашивают их по Гимзе для выявления рисунка светлых и темных полос, который является уникальным для каждой хромосомы. Каждая полоса охватывает от 5-10х106 пар оснований ДНК, которая может содержать от нескольких генов в нескольких их сотен.
Использование интерфазных ядер имеет свои преимущества, поскольку этот метод не требует митотических клеток, а хромосомы в интерфазе менее конденсированными, что позволяет получить разрешение расстояние в пределах от 50 до 500х103 оснований ДНК. Метод волоконной РІ5Н позволяет растягивать хромосомы и получать разрешение до нескольких тысяч оснований ДНК (килобейсов).
Источник
Обычно мужчины и женщины имеют хорошо выраженный фенотип, определяемый их набором хромосом — XY или XX. Но иногда рождаются дети с необычным числом половых хромосом, и это происходит в результате ненормального развития гамет. Два подобных синдрома названы по именам первых описавших их врачей. Синдром Клайнфельтера проявляется у мальчиков, которые обычно высокие, с гинекомастией (развитие молочных желез по женскому типу), пониженным умственным развитием и маленькими яичками. В 1959 году Якобе и Стронг установили, что синдром Клайнфельтера связан с наличием лишней Х-хромосомы, то есть с набором хромосом XXY.
Другой случай ненормального развития гамет называется синдромом Тернера и проявляется у девочек. У них нет яичников, они невысокие, с недоразвитыми вторичными половыми признаками (маленькая грудь). Хромосомный набор у таких девочек — Х0, то есть одна Х-хромосома (0 обозначает отсутствие хромосомы). Поскольку такие женщины гомозиготны по Х-хромосоме, у них проявляется рецессивный фенотип, например дальтонизм, обычно свойственный мужчинам. Рождение одного ребенка с генотипом XXY приходится приблизительно на каждые 700 рождений, а с генотипом ХО — на каждые 2500. Кроме того, на каждую 1000 рождений приходится один случай XXX; эти девочки внешне нормальные, хотя и с некоторыми недостатками умственного развития.
Рис. 5.6. Последствия нерасхождения Х-хромосом в ооците на первом этапе мейоза и оплодотворения сперматозоидами с хромосомами X или Y. Нерасхождение на втором этапе мейоза (не показано) может привести к еще большему увеличению числа Х-хромосом
Как же возникают такие случаи? Непосредственная причина пока еще не известна, но ясно, что во время мейоза эти хромосомы не расходятся как следует (рис. 5.6).
Такое явление и называется нерасхождением хромосом. Оно происходит в гаметах каждого пола, на первом или втором этапе мейоза или сразу на двух этапах. В результате нерасхождения образуются гаметы с двумя половыми хромосомами (XX, YY, XY) или вовсе без половой хромосомы. В редких случаях встречаются и более двух копий хромосом. (Нерасхождение аутосом приводит к серьезным врожденным порокам, о чем упоминается в гл. 14.) Если сперматозоид XY оплодотворяет яйцеклетку X, то образуется зигота с хромосомами XXY; сперматозоид без половых хромосом и яйцеклетка X дают зиготу ХО. В результате оплодотворения яйцеклетки X сперматозоидом XX получается зигота XXX, а в результате оплодотворения такой яйцеклетки сперматозоидом YY — зигота XYY.
Ненормальные случаи распределения половых хромосом заставляют задуматься, почему ХХ-женщи-ны и XY-мужчины получаются нормальными, если у них разное число Х-хромосом? Должен быть какой-то механизм, который компенсирует различие и поддерживает генетическое равновесие. В 1961 году Мэри Лион и Лайан Рассел независимо друг от друга предложили гипотезу, объясняющую компенсацию генов, связанных с Х-хромосомой. Они заметили, что у гетерозиготных женских особей часто бывает разный фенотип; гетерозиготным кошкам, например, свойственна пятнистая окраска, причем черные и желтые пятна находятся в разных местах. Лион и Рассел предположили, что в каждой клетке развивающегося эмбриона одна из Х-хромосом случайным образом «выключается», и во всех клетках, происходящих из этой клетки эмбриона, эта Х-хро-мосома продолжает оставаться в неактивном состоянии. Так, у гетерозиготной кошки в некоторых участках кожи должна быть выключена Х-хромосома с аллелем черного меха, поэтому на этих участках появляется желтый мех; на других участках кожи выключена Х-хромосома с аллелем желтого меха, и на них вырастает черный мех. Хотя такая схема деактивации Х-хромосом наиболее очевидна на примере окраски кошек и мышей, в действительности каждая женская особь млекопитающего сочетает в себе два типа клеток, и любая разница в аллелях Х-хромосом может привести к фенотипическому разнообразию.
«Выключенные» Х-хромосомы сворачиваются в плотные комочки, которые называются половыми хроматинами, или тельцами Барра, по имени открывшего их Муррея Барра; их можно увидеть в клетках нормальных женских особей. Одна из Х-хромосом остается активной, а другая сжимается, поэтому у обычной женщины тельце Барра можно обнаружить в каждой клетке, а у женщины с синдромом Тернера их нет. У женщин с лишними хромосомами их бывает два, три или даже четыре. У мужчин телец Барра обычно не бывает, но у людей с синдромом Клайнфельтера бывает одно, два и более телец Барра, в зависимости от количества лишних Х-хромосом.
Источник