
Ребенок с синдромом секкеля фото детей

Что такое синдром Секкеля?
Синдром Секкеля чрезвычайно редкое наследственное заболевание, характеризующееся задержкой роста до рождения (задержка внутриутробного развития), что приводит к низкой массе тела при рождении. Задержка роста продолжается после рождения (послеродовой период), что приводит к низкорослости (карликовость). Другие симптомы и физические особенности, связанные с синдромом Секкеля, включают аномально маленькую голову (микроцефалия); разную степень умственной отсталости; и/или необычные характерные черты лица, включая «клювоподобный» выступ носа. Другие черты лица могут включать аномально большие глаза, узкое лицо, деформированные уши и/или необычно маленькую челюсть (микрогнатия). Кроме того, у некоторых младенцев может наблюдаться постоянная фиксация пятых пальцев в согнутом положении (клинодактилия), деформация (дисплазия) тазобедренного сустава, вывих кости в предплечье (радиальный смещение) и/или другие физические отклонения.
Признаки и симптомы
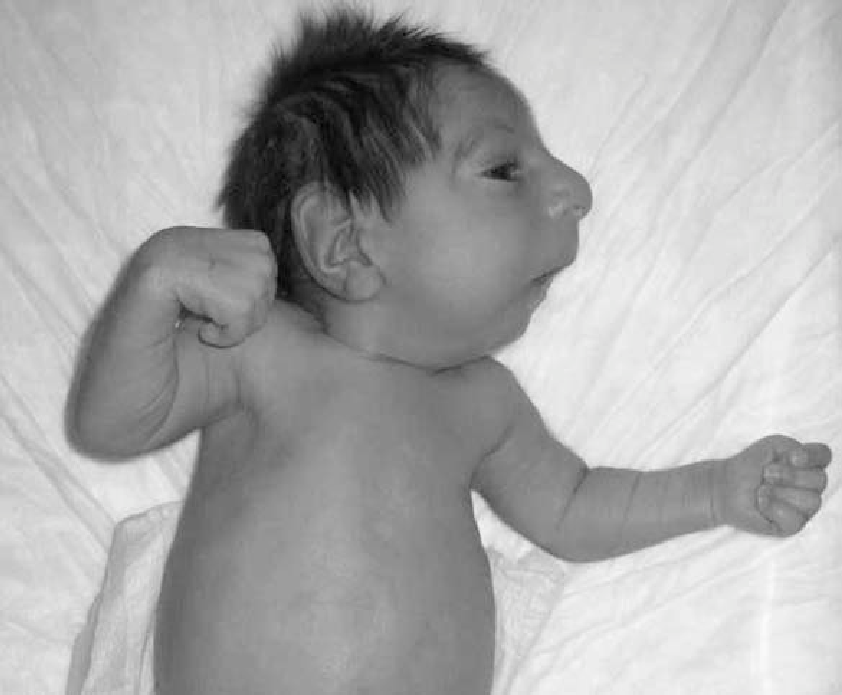
Синдром Секкеля характеризуется аномально медленным развитием плода и низкой массой тела при рождении. После рождения у ребенка будет наблюдаться медленный рост и созревание костей, что приведет к низкому, но пропорциональному росту (в отличие от карликовости коротких конечностей или ахондроплазии). Больные с синдромом Секкеля имеют различные физические отклонения и особенности развития, в том числе:
- Очень маленький размер и вес при рождении (в среднем 3,3 фунта или 1,4 кг);
- Чрезвычайно маленький, пропорциональный рост;
- Аномально маленький размер головы (микроцефалия);
- Клювовидный выступ носа;
- Узкое лицо;
- Деформированные уши;
- Необычно маленькая челюсть (микрогнатия);
- Умственная отсталость, часто тяжелая, с IQ (коэффициент интеллекта) менее 50.
Другие симптомы могут включать аномально большие глаза, высокое арочное небо, пороки развития зубов и другие деформации костей. Также часто наблюдаются такие заболевания крови, как анемия (низкий уровень эритроцитов), панцитопения (недостаток клеток крови) или острый миелоидный лейкоз (тип рака крови).
В некоторых случаях у мужчин яички не могут опускаться в мошонку (крипторхизм), а у женщин может быть ненормально увеличенный клитор. Кроме того, у больных с синдромом может быть чрезмерное оволосение на теле и одна единственная глубокая складка на ладонях (известная как обезьянья складка).
Причины
Синдром Секкеля — чрезвычайно редкое заболевание, передающееся по аутосомно-рецессивному типу. Три варианта синдрома Секкеля включают нарушения или изменения (мутации) генов на трех разных хромосомах. Расположение генной карты: синдром Секеля 1 на хромосоме 3 (3q22-q24); Синдром Секкеля 2 на хромосоме 18 (18p11.31-q11) и синдром Секеля 3 на хромосоме 14 (14q21-q22).
Хромосомы, присутствующие в ядре клеток человека, несут генетическую информацию для каждого человека. Клетки человеческого тела обычно имеют 46 хромосом. Пары человеческих хромосом пронумерованы от 1 до 22, а половые хромосомы обозначены X и Y. У мужчин одна X и одна Y хромосома, а у женщин две X хромосомы. У каждой хромосомы есть короткое плечо, обозначенное буквой «p», и длинное плечо, обозначенное буквой «q». Хромосомы далее подразделяются на множество пронумерованных полос. Например, «хромосома 3q22-q24» относится к области между полосой 22 и полосой 24 на длинном плече хромосомы 3. Подобным образом «хромосома 18p11.31-q11.2» относится к более широкой области между полосой 11p31 на коротком плече хромосомы 18 и полосе 11.2 на длинном плече хромосомы 18. Опять же, хромосома 14q21-q22 относится к более узкой области на длинном отрезке хромосомы 14 между полосами 21 и 22. Пронумерованные полосы указывают расположение тысяч генов, присутствующих на каждой хромосоме.
Специфический ген, участвующий в синдроме Секкеля 1, известен как ген атаксии-телеангиэктазии и ген Rad3-родственного белка (ATR). Гены, участвующие в синдроме Секкеля 2 и 3 типов, неизвестны.
Генетические заболевания определяются комбинацией генов определенного признака, которые находятся на хромосомах, полученных от отца и матери.
Рецессивные генетические расстройства возникают, когда человек наследует один и тот же аномальный ген одного и того же признака от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, он будет носителем болезни, но обычно бессимптомным. Риск для двух родителей-носителей передать дефектный ген и, следовательно, иметь больного ребенка, составляет 25% при каждой беременности. Риск иметь ребенка, который будет носителем, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей и будет генетически нормальным по данному признаку, составляет 25%. Риск одинаков для мужчин и женщин.
Все люди несут несколько аномальных генов. Родители, которые являются близкими родственниками (кровными родственниками), имеют больше шансов, чем неродственные родители, иметь один и тот же аномальный ген, что увеличивает риск рождения детей с рецессивным генетическим заболеванием.
Затронутые группы населения
Синдром Секкеля — чрезвычайно редкое наследственное заболевание, которое поражает в равной степени мужчин и женщин. Точная частота расстройства неизвестна. С момента первоначального описания в 1960 г. в медицинской литературе было зарегистрировано более 100 случаев.
Близкие по симптомам расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Секкеля. Сравнения могут быть полезны для дифференциальной диагностики:
Синдром Секкеля — один из шести расстройств в классе карликовости, известном как «изначальная карликовость». Эти расстройства имеют схожие характеристики, включая пороки развития скелета (дисплазию), дефицит роста до рождения (задержка внутриутробного развития) и в младенчестве и детстве, что в конечном итоге приводит к разной степени низкого роста. В эту группу заболеваний в настоящее время входят пять расстройств: синдром Секкеля, синдром Мейера-Горлина; Синдром Рассела-Сильвера; Остеодиспластическая примордиальная карликовость Маевского I/III; и остеодиспластическая примордиальная карликовость Маевского II типа.
Остеодиспластическая примордиальная карликовость Маевского II типа является чрезвычайно редким генетическим заболеванием, характеризующимся низким ростом, низкой массой тела при рождении, аномально маленькой головой (микроцефалия) и/или аномалиями скелета. Другие физические признаки могут включать большие глаза, выступающий нос в форме клюва, опущенную челюсть и/или узкое лицо. Обычно возникает серьезная недостаточность роста до рождения (внутриутробная недостаточность роста). В некоторых случаях может присутствовать умственная отсталость. Также могут появиться различные дополнительные симптомы. Конкретные симптомы и степень тяжести варьируются от случая к случаю. Считается, что II тип остеодиспластической примордиальной карликовости Маевского наследуется по аутосомно-рецессивному типу.
Диагностика
С появлением технически совершенной ультасонографии синдром Секкеля можно диагностировать до рождения (пренатально). При ультразвуковом исследовании плода используются отраженные звуковых волны для создания изображения развивающегося плода. После рождения можно заподозрить синдром Секкеля на основании тщательного клинического обследования, подробного анамнеза пациента и различных специализированных тестов. Хотя характерные черепно-лицевые, скелетные и/или другие физические аномалии, связанные с синдромом Секкеля, могут присутствовать при рождении, в некоторых случаях диагноз может не подтверждаться до тех пор, пока больной ребенок не вырастет и не разовьется полный синдром (например, когда станет очевидным низкий рост и/или умственная отсталость).
Низкий рост, связанный с синдромом Секкеля, предполагает пропорциональный рост рук и ног, что позволяет проводить дифференциальную диагностику от синдромов, связанных с низким ростом и аномально маленькими руками и ногами (карликовость с короткими конечностями).
Стандартные методы лечения
От синдрома Секкеля нет лекарства. Некоторые лекарства могут быть назначены для устранения других симптомов, связанных с заболеванием.
Лечение направлено на решение любой медицинской проблемы, которая может возникнуть, особенно заболеваний крови и структурных деформаций. Людям с умственными недостатками и их семьям потребуется соответствующая социальная поддержка и консультационные услуги.
Прогноз
Дети, страдающие синдромом Секкеля, могут жить в течение длительного периода времени, хотя они часто сталкиваются с глубокими психическими и физическими отклонениями.
Источник
Синдром Секкеля является унаследованной формой врожденной карликовости , а это значит, что младенец начинает очень мало и не может нормально расти после рождения. В то время как люди с синдромом Секкеля обычно будут пропорциональны по масштабу, у них будет отчетливо малый размер головы. Также распространена умственная отсталость.

Несмотря на множество физических и умственных проблем, с которыми сталкивается человек с синдромом Секкеля, многие из них, как известно, хорошо живут более 50 лет.
Причины синдрома Секкеля
Синдром Секкеля является наследственным расстройством, связанным с генетическими мутациями на одной из трех разных хромосом. Считается, что это бывает крайне редко, с немногим более 100 случаев, зарегистрированных с 1960 года. Многие дети, получившие диагноз синдрома Секкеля, родились у родителей, которые тесно связаны (родственники), например, брак с кузенами или братьями и сестрами.
Синдром Секкеля является рецессивным генетическим заболеванием, он возникает только тогда, когда ребенок наследует один и тот же ненормальный ген от каждого родителя. Если ребенок получает один нормальный ген и один ненормальный ген, ребенок будет носителем синдрома, но обычно не проявляет симптомов.
Если оба родителя имеют одинаковую хромосомную мутацию для синдрома Секкеля, их риск иметь ребенка с синдромом Секкеля составляет 25%, а риск наличия носителя — 50%.
Характеристики синдрома Секкеля
Синдром Секкеля характеризуется аномально медленным развитием плода и низким весом при рождении.
После рождения у ребенка будет медленный рост и созревание костей, что приводит к короткому, но пропорциональному росту (в отличие от ахондроплазии). Лица с синдромом Секкеля обладают различными физическими и характеристиками развития, в том числе:
Очень маленький размер и вес при рождении
- Чрезвычайно малый, пропорциональный рост
- Аномально небольшой размер головы (микроцефалия)
- Клювообразный выступ носа
- Узкое лицо
- Нарушенные уши
- Необычно маленькая челюсть (микрогнатия)
- Умственная отсталость, часто тяжелая с IQ менее 50
Другие симптомы могут включать аномально большие глаза, высокое арочное небо, повреждение зубов и другие деформации кости. Расстройства крови, такие как анемия (низкие эритроциты), панцитопения (недостаточно клеток крови) или острый миелоидный лейкоз.
В некоторых случаях семенники у мужчин не спускаются в мошонку, а женщины могут иметь аномально увеличенный клитор. Кроме того, у людей с синдромом Секкеля могут быть чрезмерные волосы на теле и единая глубокая складка на ладонях (известная как складки обезьян).
Диагностика синдрома Секкеля
Диагноз синдрома Секкеля основан почти исключительно на физических симптомах. Возможно, потребуется рентгеновское изображение и другие инструменты (МРТ, КТ), чтобы отличить его от других подобных условий. В настоящее время нет лабораторных или генетических тестов, специфичных для синдрома Секкеля. В некоторых случаях окончательный диагноз не может быть сделан до тех пор, пока ребенок не станет старше и не появятся характерные симптомы.
Лечение синдрома Секкеля
Лечение синдрома Секкеля сосредоточено на любой медицинской проблеме, которая может возникнуть, особенно при нарушениях крови и структурных деформациях. У людей с психическими расстройствами и их семьям должна быть предоставлена соответствующая социальная поддержка и консультационные услуги.
Источник
У моей шестилетней дочки недавно диагностировали синдром Шерешевского-Тернера. Никак не могли найти причину, почему не растет, и вот наконец докопались до истины (до этого пять лет противоположный по кариотипу диагноз стоял). Главный эндокринолог города сказала, что ГР нужен однозначно и синдром в перечне показаний, но без инвалидности не могут давать ГР. И сама же сказала, что инвалидность тоже не дадут — кроме роста проблем со здоровьем пока не диагностировали. Записаны на полное обследование, ложимся через три недели. Но может сейчас кто расскажет-подскажет, как поступают в подобных ситуациях? Пока перспективы очень грустные ((((((((( А барышня очень маленькая и уже конкретные проблемы из-за роста огребаем по полной программе
Ниночка, добрый день. У меня есть контакт семьи девочки, с таким же диагнозом. Я с ними созвонюсь, дам Ваши данные.. Еще точно видела в очереди на комиссии по назначению гормона роста маму с девочкой с этим синдромом, т.е. скорее всего они его тоже бесплатно получают, но точно не скажу. И мы из Беларуси.
Ниночка, здравствуйте! Может эту группу назвать СИДРОМАЛЬНЫЕ ФОРМЫ НИЗКОРОСЛОСТИ, тк я думаю, у многих с недостатком ГР имеется синдром. В ЭНЦ перечислены синдромы низкорослости: ШЕРЕШЕВСКОГО-ТЕРНЕРА, НУНАНА, ААРСКОГО, ЛАРОНА, СИЛЬВЕРА-РАССЕЛА, ПРАДЕРА-ВИЛЛИ, СЕККЕЛЯ. Думаю родители таких деток захотят найти таких же, с таким же синдромом и пообщаться.
Мне хочется найти родителей у кого были или есть детки с синдромом СЕККЕЛЯ. Оч мало инфо о них, слышала, что мало живут. Что с ними происходит? У меня ребёночек син. Секкеля нам собираются назначить ГР.
Ольга, у большинства участников группы как раз тот дмагноз, который обозначен в названии группы. Кроме того, взрослых здесь больше, чем родителей детей с ГН (чем бы она ни была вызвана).
Можно было бы создать здесь тему “ГЕНЕТИКА”, она касалась бы проблем детей (и взрослых) с генетически обусловленной низкорослостью.
Воообще, конечно, сложно найти людей со схожими проблемами – а как хотелось бы делиться информацией, задавать вопросы, высказывать опасения….
Предлагаю еще, если найдете сообщества с подобной тематикой, скиньте сюда ссылки! Так всегда мало инфы и много непонятного, лишним не будет.
Татьяна, генетически обусловленная низкорослость – это генетически обусловленный дефицит соматотропного гормона. Это могут быть как изученные синдромы, так и не изученные, т.о. все, кто рожден с дефицитом гр (а не приобрел его в результате операции на гипофизе либо не имеет аденомы гипофиза, подавляющей выработку стг) могут себя зачислить к теме “генетика”? П.с. Кому делали анализ на PIT1 (PROP1)? Кто из родителей детей с дефицитом ГР (любой формы ген.мутации) планировал еще ребенка? Делали ли анализы сами родители?
Добрый день!хотелось бы поделиться своей историей про генетику, вдруг кому-нибудь пригодится.Моему сыну 2,3. Родился маленький 47 см.В месяц была операция-порок сердца.ну а дальше росли мы по чуть-чуть, все врачи твердили, что с ребенком все в порядке, ездили в генетический центр, нам там даже кровь не стали брать. Спасибо этой группе, списалась с девушкой Марианной, она посоветовала хорошего эндокринолога, та в свою очередь созвонилась с генетиком. Сдали кровь, все в порядке, кариотип нормальный.Сдали кровь на fish на субтеломерные отделы, вот тут и всплыла наша бяка;((((выявили делецию субтеломерного локуса длинного плеча хромосомы 12.Бяка эта редкая, примеров ни в России ни за границей наш генетик не нашла. В итоге собираемся весной оформлять инвалидность по генетике, дабы дали нам этот заветный гормон роста. У сына прибавка роста хорошая и видимо нам он будет назначен временно для того,чтобы нагнать сверстников.Сумбурно написала, но как получилось;)
Диана, здравствуйте! Спасибо за Вашу историю! Я тоже хочу еще раз проконсультироваться с генетиком, если можно, дайте, пожалуйста, контакты Вашего врача – ФИО, где работает (можно в личку). Хочется найти хорошего генетика, занимающегося такой тематикой, имеющего дело с редкими генетическими отклонениями.
С эндокринологами я общалась в ГПМА, нас сейчас ведет Скородок Ю.Л.
Здравствуйте,Татьяна. Не за что. Эндокринолог у нас тот же. Генетик у нас Лязина Лидия Викторовна.Принимает в Медико-генетическом центре на Тобольской д5. Телефон 2947000. Прекрасный специалист, а главное человек. Сколько стоит платный прием подсказать не могу, мы по направлению наблюдаемся.
А скажите пожалуйста анализ на генетические отклонения только генетик может назначить! И как они решают кому назначать,а кому нет’
Мария, по сути вы сами можете сдавать анализы,платно.Я где-то читала,но врать не буду.На каком-то форуме читала,что это удовольствие дорогое.
Мы были у генетика и она нам сказали по внешнему виду ни каких отклонений по генетике не видно! И ни чего нам не назначила! Вот как мне убедить врача,чтоб сдать анализы!
Мария, ну вот у нас в первый раз все точно так же было:все нормально,идите гуляйте
И как вы заставили вам сделать направление или нашли другого генетика
Мария, нет,направление у нас было от педиатра (мы его сами попросили),думаю с этим проблем быть не должно.просто попали к другому генетику.
Диана, спасибо за информацию!попробую найти другого генетика!
Мы на нордитропине сейчас три месяца как. Прирост 3,5 см за это время. Корытко ооочень довольна результатом. Но нордитропина больше в городе нет и в Москве тоже и все отвечают, что до нового года его поставок не будет. Пока не знаю, что дальше делать, какой другой препарат покупать. Инвалидность нам не дали — прошли все этапы МСЭ, в том числе Москву. Ответ, что дадут только при неэффективности гормонозаместительной терапии. В новом приказе 664-н нет генетических заболеваний вообще, так что можем пройти только по этой статье — низкорослость. Так что гормон покупаем сами (помогает благотворительная организация). Но больше чем еще на три месяца они не смогут нам помочь, все фонды, что лечить в данном случае должно государство.
Мария, нам наш диагноз заподозрила тоже Лязина и назначила фиш, хотя стоял противоположный диагноз. Но она осмотрела и сказала, что по всем признакам ШТ
Источник
Автор:
07 апреля 2017 16:46
Каждый родитель надеется, что его ребенок родится на свет здоровым. Но иногда генетический механизм дает сбой, и на свет появляются младенцы с отклонениями, приводящими в изумление и ужас даже медиков. Эти дети становятся знаменитостями в мире медицины – врачи изучают их пороки, чтобы понять, как избавить от них будущие поколения. Вот самые известные подобные случаи.
Мальчик с тремя руками

Источник:
Полимелия – редкий (один случай на миллион родившихся) генетический дефект, при котором ребенок рождается на свет с лишними конечностями. Самый недавний и известный случай полимелии произошел в 2014 году в Индии, где родился мальчик с двумя левыми руками. Ребенку повезло: врачи, несмотря на риск, связанный с операциями у младенцев, быстро удалили лишнюю конечность. Мальчик благополучно оправился после операции.
Киран Вейтц

Источник:
Эта девочка родилась с редчайшим дефектом – Ectopia Cordis, или внешнее расположение сердца. Сердце Киран расположено вне тела. Усугубляется это тем, что и печень у девочки находится снаружи – невероятно редкое сочетание! Как правило, такие дети не живут и года, но благодаря тщательному уходу и медицинскому наблюдению Киран живет и неплохо себя чувствует. Недавно ей исполнилось два года.
Аршид Али Хан

Источник:
Индийский мальчик Аршид Али Хан родился с редкой генетической патологией – с настоящим хвостом. Впрочем, это стало для него настоящим благословением: жители деревни считали его избранным, поклонялись ему и называли “Богом-Обезьяной”. Правда, по этой причине они бурно препятствовали родителям мальчика, которые хотели избавить сына от атавизма. В итоге, операцию по удалению хвоста мальчику сделали только в 14 лет.
Сэм Бернс

Источник:
Сэм Бернс с рождения страдал прогерией – ускоренным старением всех систем организма. На этом снимке, на котором он выглядит довольно пожилым мужчиной, Сэму 10-12 лет. Сэм умер от старости в 17 лет, успев отдать немало времени и сил просвещению населения по вопросам прогерии и поддержке людей с аналогичной болезнью.
Габриэль Мейсон

Источник:
Габриэль Мейсон родился с синдромом Протеуса – болезнью, которая характеризуется ускоренным ростом кожи и костной ткани. Если вы смотрели фильм Дэвида Линча “Человек-слон”, вы представляете, как выглядит человек, страдающий этой болезнью: именно она была у главного героя. Первую операцию по удалению разросшихся костей стоп Габриэлю сделали в 18 месяцев. Теперь ему предстоят операции каждые полгода, чтобы скорректировать рост тканей и дать ребенку возможность научиться ходить.
Руна Бегум

Источник:
Руна Бегум из Индии родилась с экстремально выраженной гидроцефалией – скоплением жидкости в полости черепа вокруг головного мозга. Окружность ее головы близилась к метру, когда врачи решили, несмотря на риск, делать операцию. Теперь ее голова стала заметно меньше, и все-таки при окружности почти в 70 сантиметров выглядит, откровенно говоря, ужасно. Тем не менее, девочке повезло, как минимум, дважды: она выжила после рождения, и врачи сумели оперативным вмешательством улучшить ее состояние. Что с ней будет дальше, неизвестно.
Бренна Уэстлейк

Источник:
Бренна Уэстлейк родилась с ихтиозом арлекина – состоянием, при котором кожа растет чрезмерно активно, покрываясь язвами и трещинами и со временем становясь похожей на слоновью. Больные с ихтиозом арлекина лишь в половине случаев доживают до года, а процент переживших 18-летний рубеж и вовсе невелик. Врачи не знают средства от этой болезни, так что прогнозы на будущее у Бренны совсем не радужные.
Анна Рашель Пондэр

Источник:
Анна Рошель Пондэр – еще один знаменитый случай прогерии. Девушка дожила до 18 лет, при этом врачи говорили, что к этому нежному возрасту ее организм соответствовал 144 годам.
Сонг Шенг

Источник:
Сразу после рождения кожа мальчика из китайского города Цзиньху начала покрываться чешуйками. Врачи диагностировали у него чешуйчатый ихтиоз, при котором кожа, неспособная поддерживать правильный температурный режим, нарастает слоями, образуя, в итоге, толстый слой ороговевших чешуек, похожих на рыбьи. Помимо кожного дефекта, из-за отсутствия нормальной температурной регуляции больной страдает от постоянно повышенной температуры. Лечения от этой болезни пока нет.
Анджела Моралес

Источник:
Анджела Моралес родилась с анэнцефалией – грубым пороком развития, при котором у младенца недоразвиты или полностью отсутствуют большие полушария мозга, а также верхняя часть костей черепа. Как правило, обнаружив этот порок во время беременности, врачи советуют родителям сделать аборт, поскольку такие младенцы обычно не в состоянии прожить и дня. Однако родители Анджелы отказались убить нерожденного ребенка, а после рождения так тщательно ухаживали за ней, что девочка прожила год и продолжает жить. Возможно, она станет для врачей ключом к решению проблемы анэнцефалии.
Супатра Сасупхан

Источник:
11-летняя Супатра Сасупхан из Индии попала в Книгу рекордов Гиннеса как “самая волосатая девочка в мире”. Она родилась с редкой патологией – синдромом Амбраса, или гипертрихизмом, при которой волосы растут по всему телу. Врачам пришлось даже сделать Супатре операцию, чтобы дать ей возможность дышать. Несмотря ни на что, Супатра с редким оптимизмом смотрит на мир. Синдром Амбраса – чрезвычайно редкое заболевание: во всем мире врачам известно лишь 50 случаев этой болезни.
Лакшми Татма

Источник:
Лакшми Татма из Индии родилась с редчайшей формой полимелии: у нее было четыре руки и четыре ноги! 27 часов бригада медиков из 30 человек проводила сложнейшую операцию по удалению у девочки лишних конечностей. Операция прошла удачно, и теперь Лакшми вполне здорова.
Шилох Пепин

Источник:
Шилох Пепин родилась с редким дефектом – сиреномелией. ее ноги были сросшимися в нижней части. образуя подобие рыбьего хвоста. О ней много писали в прессе, называя “первой земной русалкой”. К несчастью, из-за неправильного развития конечностей у Шилох неправильно сформировалось большинство внутренних органов. Девочка за свою жизнь пережила около 150 операций и умерла в 10-длетнем возрасте от пневмонии.
Манар Могед

Источник:
Воистину жуткое состояние, в котором родилась Манар Могед, называется паразитарной краниопагией. При этой патологии к голове ребенка в утробе матери прирастает его недоразвившийся близнец или отдельная часть его тела, как правило, голова. Врачи наблюдали девочку до 10-месячного возраста, после чего в ходе 16-часовой операции избавили ее от нежизнеспособного паразитического организма.
Циклопия

Источник:
Циклопия – известный медикам, оно очень редкий дефект, при котором ребенок рождается с одним глазом. Как правило, циклопия сопровождается другими грубыми пороками развития. В результате ребенок с этим дефектом обычно умирает еще в утробе матери или сразу после рождения. Этот младенец в Египте, однако, родился живым и прожил достаточно долго, чтобы врачи сумели познакомиться с этой редчайшей патологией. Впрочем, больше нескольких дней такие дети не способны прожить ни при каких обстоятельствах.
Источник: — переведено специально для fishki.net
Ссылки по теме:
Понравился пост? Поддержи Фишки, нажми:
Источник