
Синдром блоха сульцбергера мкб 10

Содержание
- Описание
- Дополнительные факты
- Причины
- Симптомы
- Диагностика
- Лечение
Названия
Название: Синдром Блоха-Сульцбергера.
Синдром Блоха-Сульцбергера
Описание
Синдром Блоха. Сульцбергера – наследственная форма нарушения пигментации кожи, которая часто сочетается с пороками развития зубов, волос, ногтей и глаз. Симптомы заболевания характеризуются выраженной стадийностью – сначала на коже появляется эритематозная сыпь в виде пятен и линий, затем на ее месте развивается гиперкератоз, сменяющийся пятнами и последующей гипопигментацией с атрофией кожных покровов. Диагностика синдрома Блоха-Сульцбергера производится на основании данных настоящего статуса больного, гистологического исследования образцов кожи в области поражения, изучения наследственного анамнеза и молекулярно-генетических анализов. Специфического лечения этой патологии на сегодняшний момент не существует, используют симптоматические и поддерживающие мероприятия различного характера..
Дополнительные факты
Синдром Блоха-Сульцбергера (семейная форма недержания пигмента, нейрокожный меланобластоз) – генетическое заболевание, характеризующееся нарушением метаболизма меланина в коже и рядом сопутствующих пороков развития. Впервые это заболевание было описано в 1926 году швейцарским дерматологом Б. Блохом, затем более детальное изучение данной патологии провел американский педиатр М. Сульцбергер в 1929 году и, независимо от предыдущих исследователей, немецкий врач Г. Сименс. Именно поэтому в литературе можно найти другое название этого заболевания – синдром Блоха-Сименса. Удалось выяснить, что патология наследуется сцепленно с Х-хромосомой, при этом мутантный аллель является доминантным. По этой причине синдром Блоха-Сульцбергера во много раз чаще встречается у девочек – половое распределение составляет примерно 6:210, так как наличие этой мутации у эмбрионов мужского пола практически всегда является летальным и приводит к самопроизвольному прерыванию беременности. Развитие заболевания у мальчиков может быть обусловлено генетическим мозаицизмом, наличием сопутствующего синдрома Клайнфельтера или редких точечных «мягких» мутаций. Общая встречаемость синдрома Блоха-Сульцбергера составляет примерно 1 случай на 75 000 новорожденных..
Синдром Блоха-Сульцбергера
Причины
При синдроме Блоха-Сульцбергера происходит повреждение гена IKBKG, который располагается на Х-хромосоме. Продуктом его экспрессии является многофункциональный сложный белок – регуляторная субъединица NEMO-ингибиторной киназы, участвующей в сигнальной системе важного транскрипционного фактора (NF-каппа-B). Этот фактор и соответствующий ему сигнальный путь регулирует огромное количество различных процессов в организме человека – участвует в процессах адаптации при стрессе, иммунном ответе, некоторых формах воспалительных реакций, процессах клеточной адгезии, а также тормозит процессы апоптоза. Наиболее часто причиной синдрома Блоха-Сульцбергера становятся крупные транслокации и делеции гена IKBKG, в результате чего экспрессия и выделение белка с этого гена полностью прекращаются..
Поскольку у женщин имеется две Х-хромосомы, при наличии второй нормальной аллели гена IKBKG такая мутация не угрожает жизни, но обуславливает развитие синдрома Блоха-Сульцбергера. Известен факт, что в соматических клетках женского организма всегда активна только одна Х-хромосома, тогда как вторая сконденсирована в половой хроматин. Значительная вариабельность выраженности симптомов заболевания обусловлена распределением клеток, где активна хромосома именно с мутантной формой гена IKBKG. Как следствие, в вышеуказанных клетках не образуется регуляторной субъединицы NEMO-ингибиторной киназы, что и приводит к характерным порокам развития, формирующим клиническую картину синдрома Блоха-Сульцбергера. Кожные симптомы связаны с нарушением проницаемости мембран меланоцитов (в результате чего практически весь пигмент беспрепятственно покидает клетки) и аутоиммунными реакциями..
В отличие от женщин, у мужчин в норме есть лишь одна Х-хромосома, поэтому при наличии нонсенс-мутации в гене IKBKG выделение важного белка не происходит абсолютно во всех клетках организма. Это становится причиной массированного апоптоза гепатоцитов еще на этапе внутриутробного развития – в норме этот процесс задерживается как раз системой NF-каппа-B. Развитие нарушений, подобных синдрому Блоха-Сульцбергера, у мальчиков возможно при наличии сопутствующего синдрома Клайнфельтера (кариотипа XXY) или генетического мозаицизма, когда только часть клеток в организме имеет дефект гена IKBKG. В последние годы были выявлены точечные мутации этого гена, которые не приводят к полной остановке транскрипции, но изменяют структуру конечного белка. Однако чаще всего у мальчиков с такими дефектами возникает не синдром Блоха-Сульцбергера, а другие генетические заболевания – эктодермальные дисплазии, иммунодефициты, пороки развития скелета..
Симптомы
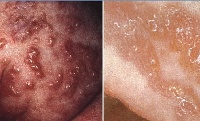
Одним из наиболее выраженных и распространенных проявлений синдрома Блоха-Сульцбергера является дерматоз, который обнаруживается при рождении или (реже) возникает на протяжении первых дней жизни новорожденного. В развитии изменений кожных покровов при этой патологии наблюдается характерная стадийность, что также является важным диагностическим признаком. Локализация таких изменений – на боковых поверхностях конечностей, туловища, шеи, вдоль линий Шарко или проекций основных нервных стволов. В большинстве случаев выделяется четыре основных стадии кожных симптомов синдрома Блоха-Сульцбергера:
1. Я стадия – воспалительная или везикулобуллезная. Начинается при рождении больного или на протяжении 2-3 недель жизни и длится до возраста 3-8 месяцев. На этом этапе заболевания возникают везикулы, эритематозная сыпь, возможно развитие пузырей и пустул. С учетом возраста больных синдромом Блоха-Сульцбергера существует определенный риск инфекционных осложнений на пораженных участках кожи..
2. Я стадия – гипертрофическая или веррукозная. Характеризуется развитием на пораженных участках тела гиперкератоза в виде бляшек, бородавчатых и лихеноидных разрастаний. Их распределение, как правило, симметричное и линейное, вдоль линий Шарко или проекций нервных стволов. Длительность этой стадии синдрома Блоха-Сульцбергера составляет несколько месяцев (до возраста одного года), у некоторых больных может отсутствовать..
3. Я стадия – пигментная. На этом этапе заболевания у больных на пораженных участках кожи возникают очаги гиперпигментации различных форм и размеров темно-коричневого цвета. Почти у половины пациентов с синдромом Блоха-Сульцбергера такие очаги появляются на неизмененных участках тела и не связаны с высыпаниями, характерными для предыдущих стадий. Длительность гиперпигментации составляет несколько лет, обычно – до периода полового созревания..
4. Я стадия – атрофическая. Характеризуется потерей пигмента на очагах поражения с развитием признаков атрофии кожи. У некоторых больных синдромом Блоха-Сульцбергера такие проявления могут быть выражены очень слабо, в отдельных случаях симптомы заболевания полностью исчезают после завершения полового созревания..
Диагностика
Для определения синдрома Блоха-Сульцбергера используют множество диагностических методов и техник – дерматологический осмотр, изучение наследственного анамнеза, гистологическое исследование пораженных участков кожных покровов, молекулярно-генетические анализы. При осмотре выявляются разнообразные (в зависимости от возраста больных и стадии заболевания) изменения кожи эритематозного, везикулярного или гиперкератического характера, у старших пациентов может определяться очаговая гипер- или гипопигментация кожи. Помимо этих проявлений, при синдроме Блоха-Сульцбергера возможна дистрофия ногтей, алопеция, аномалии строения зубов..
Наследственный анамнез может выявить семейный, доминантный и сцепленный с Х-хромосомой характер наследования патологии. В некоторых случаях у матери больной в анамнезе отмечается несколько случаев самопроизвольного прерывания беременности – это связано с внутриутробной смертью плода мужского пола. Результаты гистологического исследования тканей кожи при синдроме Блоха-Сульцбергера зависят от стадии заболевания – на первом этапе обнаруживается спонгиоз, развитие эпидермальных пузырей, заполненных эозинофилами и фибриновыми массами. На второй стадии патологии выявляются признаки внутриэпителиальной кератинизации, акантоз и гиперкератоз, в дерме отмечается отек с нейтрофильной и эозинофильной инфильтрацией. На третьей стадии синдрома Блоха-Сульцбергера воспалительные изменения в дерме (отек, инфильтрация) исчезают, но наблюдается значительное накопление пигмента в верхних слоях кожи. Четвертая стадия характеризуется исчезновением пигмента, развитием фиброзной ткани и частичным исчезновением придатков кожи..
Молекулярно-генетическая диагностика синдрома Блоха-Сульцбергера выполняется врачом-генетиком и может быть произведена несколькими основными техниками. Прямое автоматическое секвенирование последовательности гена IKBKG позволяет выявить практически любые изменения в его структуре. Транслокации и делеции значительных участков гена, часто выступающие в качестве причины синдрома Блоха-Сульцбергера, можно обнаружить при помощи методики FISH-анализа. Данное заболевание также может быть подтверждено посредством исследования инактивации Х-хромосом в клетках пораженных тканей..
Лечение
Специфического лечения синдрома Блоха-Сульцбергера на сегодняшний момент не существует, кожные поражения на воспалительном этапе заболевания обрабатываются антисептическими средствами и растворами для предотвращения инфекционных осложнений. Кроме того, рекомендуется местное назначение глюкокортикоидных стероидов для уменьшения воспаления, однако такое лечение следует производить с осторожностью, учитывая высокую проницаемость кожных покровов у детей младшего возраста. Другие проявления синдрома Блоха-Сульцбергера (пороки развития зубов, глаз) лечат при наличии показаний..
Источник
Рубрика МКБ-10: Q82.3
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q80-Q89 Другие врожденные аномалии пороки развития / Q82 Другие врожденные аномалии пороки развития кожи
Определение и общие сведения[править]
Недержание пигмента
Синонимы: Incontinentia pigmenti, синдром Блоха-Сульцбергера, синдром Блоха-Сименса
Системное заболевание, проявляющееся своеобразными стадийными изменениями кожи в сочетании с патологией глаз, зубов, волос и ногтей, ЦНС и костно-мышечного аппарата.
Эпидемиология
Наследование носит Х-сцепленный доминантный характер. Подавляющее большинство случаев семейные.
Распространенность составляет приблизительно 1 / 143,000. Соотношение женщин и мужчин составляет 20: 1.
Этиология и патогенез[править]
Недержание пигмента вызывается семейной (10-25%) или спорадической de novo (> 50%) мутацией NF-kappaB естественного модулятора гена IKBKG (ранее NEMO). 4-10 делеции экзона лежат в основе 80% случаев заболевания.
Клинические проявления[править]
Заболевание существует с рождения или проявляется в первые недели жизни. Характерна стадийность кожных изменений.
• Стадия I (пузырьковая, или воспалительная). Она возникает в первые часы жизни в виде отечной эритемы с везикулобуллезными, реже уртикарными элементами, которые склонны к линейному расположению. Содержимое пузырьков прозрачное, в нем обнаруживают эозинофилы. При вскрытии и подсыхании пузырьков образуются мелкие эрозии и корочки. Общее состояние ребенка обычно не нарушено, в периферической крови – эозинофилия.
• Стадия II (веррукозная). Она развивается приблизительно через 2-3 мес и характеризуется лентикулярными ороговевающими папулами, расположенными преимущественно линейно в зоне бывших везикул или беспорядочно, напоминая бородавчатый невус. Веррукозные изменения кожи сохраняются в течение нескольких месяцев. На ладонях и подошвах может быть диффузный гиперкератоз.
• Стадия III (гиперпигментация). Она наступает через 3-6 мес от начала заболевания. На месте регрессировавших очагов развивается пигментация коричневатого или темно-серого цвета, напоминающая брызги грязи на светлом фоне, в виде полосок и завихрений.
• Стадия IV (гипопигментация). Она проявляется на 2-3-м десятилетии жизни. Пигментация у части больных сменяется депигментацией, умеренно выраженной атрофией, очаговым склерозом.
Стадийность наблюдают не всегда. Более 10% больных имеют только пигментацию. Может быть только изолированная воспалительная стадия. Признаки различных стадий могут существовать одновременно.
Из внекожных изменений наиболее часто возникают нарушения со стороны нервной системы и органа зрения.
Недержание пигмента (incontinentia pigmenti): Диагностика[править]
Типичные поражения кожи и генетическое тестирование являются достаточными для установления диагноза.
Наблюдается также лейкоцитоз и эозинофилия. Гистологические изменения зависят от стадии заболевания.
Дифференциальный диагноз[править]
Этап I может быть неправильно диагностирован как буллезный импетиго, врожденный буллезный эпидермолиз, герпес или ветряную оспу.
Дифференциальная диагностика II стадии включает в себя бородавки, контагиозный моллюск и синдром эпидермального невуса.
Любое состояние с линейной и окружной пигментацией пересекается с клиникой стадии III.
Стадия IV напоминает рубцевание, витилиго, гипомеланоз Ито или другие гипопигментозы с локализованной алопецией.
Дополнительно следует рассматривать синдром Нейджели-Франческетти-Ядассона и болезнь Норри.
Недержание пигмента (incontinentia pigmenti): Лечение[править]
Лечение симптоматическое. На ранних стадиях назначают 10% мазь цинка оксида, мази с глюкокортикоидами, анилиновые красители, при инфицировании – антибиотики. При развитии массивных веррукозных изменений используют изотретиноин.
Прогноз
Ожидаемая продолжительность жизни в норме. Лица, не имеющие аномалий ЦНС, как правило, имеют нормальное физическое и умственное развитие.
Профилактика[править]
Недержание пигмента наследуется Х-сцепленно доминантно. Заболевшая женщина имеет 50% риск передачи патологии потомству. Больные мужского пола должны быть проверены на 47, XXY кариотип.
Прочее[править]
Источники (ссылки)[править]
Дерматовенерология [Электронный ресурс] / под ред. Ю. С. Бутова, Ю. К. Скрипкина, О. Л. Иванова – М. : ГЭОТАР-Медиа, 2013. – https://www.rosmedlib.ru/book/ISBN9785970427101.html
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Медицинский эксперт статьи
х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Недержание пигмента (меланобластоз Блоха-Сульцберга) – системное эктомезодермальное заболевание, наследуемое доминантно, Х-сцеплено с летальным эффектом для эмбрионов мужского пола.
[1], [2], [3]
Код по МКБ-10
Q82.3 Недержание пигмента [incontinentia pigmenti]
Причины и патогенез недержания пигмента
Недержание пигмента обусловлено мутантным доминантным геном, локализованным в Х-хромосоме. Ген летален для плода мужского пола. В основном (90-95 %) болеют женщины, а заболевание мужчин расценивают как результат спонтанной мутации.
[4], [5], [6]
Гистопатология меланобластоза Блоха-Сульцберга
Гистологически для первой стадии характерено образование пузырьков, содержащих эозинофилы. В эпидермисе между везикулами отмечаются единичные дискератотические клетки. В дерме обнаруживают инфильтраты, состоящие из лимфоцитов и эозинофилов. Для второй стадии характерны акантоз, нерегулярно – папилломатоз и гиперкератоз, наличие многочисленных дискератотических клеток. В базальном слое – вакуолизация клеток и уменьшение содержания в них меланина. В дерме определяется средней выраженности хронический воспалительный инфильтрат с небольшим количеством мелонофагов, проникающий во многих местах в эпидермис. Для третьей стадии характерно недержание пигмента. Отмечается проникновение пигмента в дерму и накопление его в меланофагах.
Патоморфология меланобластоза Блоха-Сульцберга
Морфологические изменения в эпидермисе отражают стадии заболевания. В I стадии отмечается спонгиоз с образованием пузырей, содержащих нейтрофильные и эозинофильные гранулоциты, фибрин. Между пузырями могут находиться дискератотические клетки. Во II стадии – гиперкератоз с большим числом дискератотических клеток, акантоз, папилломатоз, вакуольная дистрофия базальных эпителиоцитов, большое количество пигмента в базальном слое. В дерме отмечаются отек, инфильтраты из лимфоцитов, гистиоцитов. нейтрофильных и зозинофильных гранулопитов. В веррукозных элементах отмечаются псориазоформный акантоз, гиперкератоз, очаговый паракератоз, в дерме – инфильтраты из лимфоцитов, плазматических клеток, меланофагов. По мере формирования пигментных пятен (III стадия) пузыри исчезают, воспалительные изменения уменьшаются, в верхней части дермы много меланофагов. В IV стадии выявляются зоны истончения эпидермиса, очаговый гиперкератоз, уменьшение количества меланина в базальном слое эпидермиса, в сетчатом слое дермы располагается небольшое число меланофагов. При электронно-микроскопическом исследовании кожи обнаруживают повышение активноности меланогенеза в I-II стадиях процесса. Меланоциты имеют много отростков, иногда проникающих в дерму через базальную мембрану. В шиповатом слое выявлена вторая популяция меланоцитов. В стадии пигментации в дерме определяется большое количество меланофагов, нагруженных пигментом, меланоциты менее активны, содержат аутофагосомы. В эпителиоцитах нарушен транспорт меланина. В IV стадии меланоциты неактивные, они округлой формы, без длинных отростков. Число меланофагов и дерме уменьшено.
Гистогенез недержания пигмента
В основе заболевания лежит нарушение синтеза и транспорта меланина меланоцитами. В начале процесса меланогенез усилен, в последующих стадиях он заметно снижается, и в IV стадии процесса меланоциты функционально полностью истощены, а накопленный в дерме пигмент постепенно резорбцируется. Отмечается нестабильность хромосом. Предполагается, что ген локализован в участке Хр11.2. Вероятно, заболевание развивается вследствие делеции. В отличие от классического варианта ген, обусловливающий гипомеланоз Ито, расположен на хромосоме 9-9q-33qter. Возможна роль нарушений иммунной толерантности, к связи с чем происходит аутоиммунная атака на клоны клеток эктодермального происхождения, имеющие аномальные поверхностные антигены, или же имеет место преждевременная гибель дефектных клонов. Хемотаксис эозинофилов в очаги и поражения, вероятно, обусловлен наличием лейкотриена В4.
Особым вариантом недержания пигмента является сетчатый пигментный дерматоз (син: синдром Фринческетти-Ядассони, дерматоз ретикулярный пигментный Негели), который проявляется обычно на 2-м году жизни, у лиц обоего пола. Отмечается аутосомно-доминаптпый тип передачи. При этом варианте заболевания без воспалительной стадии начинается стадия гиперпигментации в виде сетки или пятен, расположенных на коже живота, шеи, груди, в области кожных складок. Характерна также диффузная или точечная кератодермия ладоней и подошв. У больных отклонений в умственном и физическом развитии не отмечено.
Гипомеланоз Ито (ахроматический вариант заболевания) возникает в раннем детском возрасте, характеризуется появлением очагов щ пигментации кожи, идентичных по очертаниям и расположению участкам гиперпигментации при типичной форме недержания пигмента, но без предшествующих двух стадий процесса. Различают кожную и нейрокожную формы, которые наследуются по аутосомно-доминантному типу. При кожной форме отсутствие пигмента наблюдается в детском возрасте. При нейрокожной форме кроме расстройства пигментации отмечаются неврологические нарушения (умственная отсталость, судорожный синдром) и костные аномалии.
Дифференциальный диагноз проводят с знтеропатическим акродерматитом, синдромом Вербова, Олбрайта, гидротичсской эктодермалыюй дисплазией, в I стадии – с буллезным эпидермолизом, герпесом, эпидемической пузырчаткой нoворождениых.
Симптомы меланобластоза Блоха-Сульцберга
Заболевание развивается при рождении ребенка или в первые дни жизни. Различают несколько вариантов недержания пигмента: классический вариант Блоха-Сульцберга, сетчатый пигментный Франческети-Ядассона и гипомеланоз Ито. Классический вариант характеризуется последовательно сменяющими друг друга тремя стадиями: буллезной (воспалительной), папуло-веррукозной и пигментной.
Клиническая картина зависит от стадии процесса. Вначале, с рождения или, реже, в первые дни или недели жизни, возникают эритемато-везикулезные, папуловезикулезные высыпания, располагающиеся преимущественно на боковых поверхностях туловища и проксимальных отделах конечностей с тенденцией к полосовидному расположению (I-II стадии). Часть элементов приобретает веррукозный характер. После регрессирования высыпаний (III стадия) остается пигментация в виде характерных “брызг”, “завихрений” и полос. С течением времени гиперпигментация постепенно сменяется нерезко выраженной атрофией, склерозом и депигментацией (IV стадия). Стадийность заболевания иногда слабо выражена, одновременно могут существовать буллезные, папулезные и пигментные очаги. Нередко III стадия появляется без предшествующих симптомов. Это может быть в тех случаях, если I и II стадии прошли во внутриутробном периоде или были стертыми и остались незамеченными. Кроме изменений кожи, у большинства больных выявляются различные экто- и мезодермальные дефекты: аномалии зубов, гипотрихоз, дистрофии ногтей, изменения глаз, скелета, ЦНС К вариантам этого заболевания относят буллезный кератогенный и пигментный дерматит, или синдром Асбо-Ханзена, сетчатый пигментный дерматоз Негели, или синдром Франческетти-Ядассона, и бесцветную форму недержания пигмента – синдром Ито, что не бесспорно. Указывается на существование переходных форм.
Буллезная стадия (I) заболевания начинается на 1-2 неделе жизни ребенка и характеризуется высыпанием пузырьков и пузырей на эритемозном основании, папуло-везикулезных и уртикарных элементов. Процесс локализуется преимущественно на конечностях, боковых поверхностях туловища. Сыпь располагается линейно, симметрично или сгруппированно. Содержимое пузырьков обычно прозрачное, при их вскрытии и подсыхании образуются мелкие эрозии и корочки. Элементы сыпи возникают приступами, распространяясь на новые участки кожи. У большинства больных общее состояние обычно не нарушено. В крови обнаруживается эозинофилия.
Папуловеррукозная стадия (II) развивается приблизительно через 4-6 недель после рождения и проявляется образованием ороговевающих, гиперкератотических папул, пустул, веррукозных разрастаний, расположенных линейно в зоне бывших везикул, или беспорядочно. Эти изменения кожи сохраняются в течение нескольких месяцев. На ладонях и подошвах развивается диффузный гиперкератоз.
Пигментная стадия (III) развивается обычно через 3-6 месяцев от начала заболевания и характеризуется появлением на месте разрешившихся очагов коричневато-желтых пятен, гиперпигментации с более светлыми краями неправильных очертаний («брызги грязи»). Эти ветвистые, линейные рисунки расположены преимущественно на коже живота и, реже, конечностей. Иногда могут наблюдаться одновременно папуло-веррукозная и пигментная стадии. Со временем (15-20 лет) на месте гиперпигменгации развивается легкая атрофия, гипопигментация, выделяемая некоторыми авторами как четвертая – атрофическая стадия болезни. В этой стадии могут наблюдаться различные экзо- и мезодермальные изменения, офтальмологическая патология (косоглазие, нистагм, катаракта, атрофия зрительного нерва, отслойка сетчатки, кератигы, голубоватые склеры, аномалии пигментации радужки), неврологические изменения (припадки, эпилепсия, олигофрения, спастические параличи по типу тетра- или параплегии), заболевания внутренних органов и опорно-двигательного аппарата, дистрофия ногтей и волос.
Лечение недержания пигмента
Эффективных методов терапии нет. В первой стадии рекомендуются небольшие дозы кортикостероидов. В стадии веррукозных разрастаний эффективным является неотигазон. Наружно применяют анилиновые красители, зпителизируюшие, противовоспалительные препараты и средства, улучшающие трофику тканей.
Источник