
Синдром денди уокера на мрт

Диагностика аномалии Денди-Уокера по МРТ, КТа) Терминология: б) Визуализация: 1. Общие характеристики аномалии Денди-Уокера:
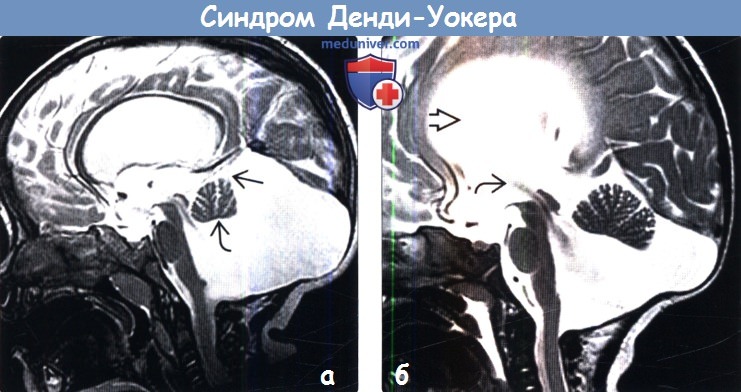
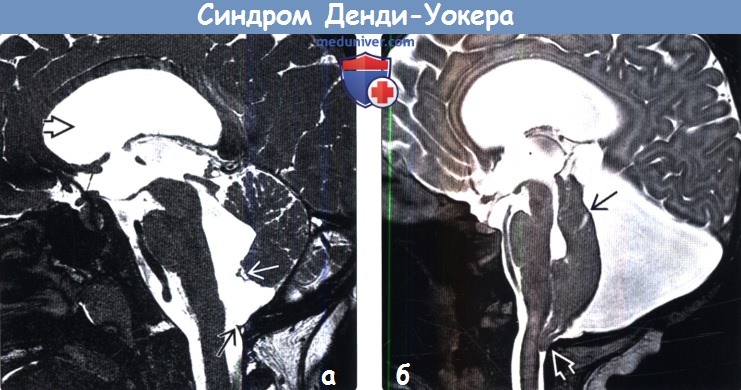
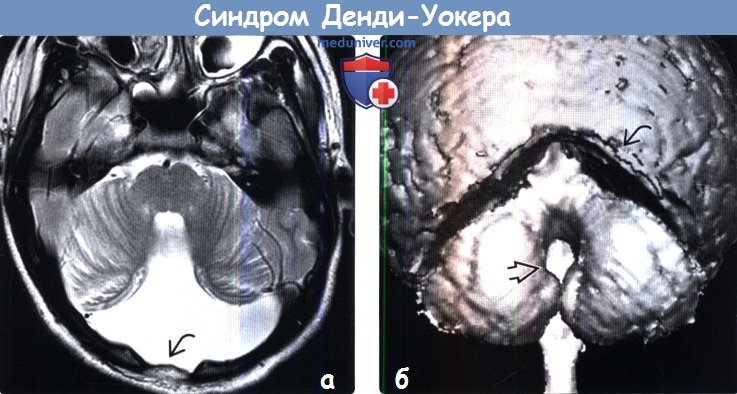
2. Рентгенологические признаки аномалии Денди-Уокера: 3. КТ при аномалии Денди-Уокера: 4. MPT при аномалии Денди-Уокера:
5. УЗИ при аномалии Денди-Уокера: 6. Несосудистые вмешательства: 7. Рекомендации по визуализации: в) Дифференциальная диагностика аномалии Денди-Уокера: 1. Спектр Денди-Уокера: 2. Арахноидальная киста (АК) задней черепной ямки: 3. Деформация «коренного зуба» (синдром Жубера): 4. Изолированные IV желудочек: г) Патология: 1. Общие характеристики: 2. Стадирование и классификация: 3. Макроскопические и хирургические особенности: 4. Микроскопия: д) Клиническая картина аномалии Денди-Уокера: 1. Проявления: 2. Демография: 3. Течение и прогноз: 4. Лечение аномалии Денди-Уокера: е) Диагностическая памятка: ж) Список литературы:
– Также рекомендуем “Ромбэнцефалосинапсис (РЭС) на МРТ” Редактор: Искандер Милевски. Дата публикации: 26.2.2019 |
Источник
Аномалия (синдром) Денди-Уокера (Dendy-Walker)-порок развития головного мозга, для которого характерна триада симптомов – гипотрофия червя мозжечка и/или полушарий мозжечка, кисты задней черепной ямки, гидроцефалия разной степени.
К сведению – пороки головного мозга 1 на 100 родов, из их 80% гидроцефалии различного генеза, в т.ч. и атрофические, из них от 9,5% до 12% – синдром Денди-Уокера (СДУ), но считается что эта цифра занижена, т.к. встречаются формы СДУ без гидроцефалии.
Этиология синдрома неизвестна. СДУ может быть проявлением генетических заболеваний, таких как синдром Меккеля (аутосомно-рецессивный путь- микроцефалия, полидактилия, поликистоз почек, глазные аномалии – микрофтальм, гипоплазия ЗН, ВПС, крипторхизм, затылочная спино-мозговая грыжа), Меккеля-Грубера (то же + энцефалоцеле), Варбурга, Тернера, трисомия 9, триплоидия, 6р- и другие хромосомные аберрации.
Предполагается, что определенную роль играют и такие факторы как вирусная инфекция (ЦМВ, краснуха), алкоголь, диабет беременной. Если синдром не связан с генетической патологией, то риск повторяемости при повторной беременности -1-5%, при наследовании по аутосомно-рецессивному типу – 25%. Сочетание с другими пороками ЦНС – 50%, особенно часто с агенезией мозолистого тела 7-17%, также часто встречаются полимикрогирия, агирия, микрогирия, пороки нижних олив.
При патологоанатомическом исследовании пороки ЦНС находят в 68% случаев с СДУ. Кроме пороков ЦНС часто встречаются ВПС, энцефалоцеле, поликистозные почки, полисиндактилия, гемангиомы, расщепления губы/неба, с Клиппеля-Фейля и др.
Впервые синдром описан Денди и Блекфаном в начале 20 века, они считали это состояние вторичным по отношению к врожденной атрезии отверстий Мажанди и Люшки, обспечивающих отток ликвора из 4 желудочка в субарахноидальное пространство; Уолкер был врачом, впервые проведшим хирургическое лечение больному с этой патологией, поэтому заболевание стало называться СДУ.
В настоящее время общепринята гипотеза, в соответствии с которой синдром представляет собой сложную аномалию развития срединных структур ромбовидного мозга. Гарднер и соавторы (1974 г.) высказали предположение , что заболевание обусловлено дисбалансом между продукцией ликвора в боковом и 3 и 4 желудочках мозга.
Ее избыточная продукция в 4 желудочке приводит к раннему расширению и выбуханию свода ромбовидного мозга, максимальное расширение на уровне 4 желудочка приводит к сдавлению и вторичной гипоплазии червя мозжечка, появлению сообщения между 4 желудочком и задней черепной ямкой и развитием кисты задней черепной ямки.
Диагностика.
Во всех случаях обнаружения кист задней черепной ямки и любой патологии задней черепной ямки, дифференциальный диагноз с арахноидальной кистой и расширением большой цистерны – для СДУ патогномоничен дефект мозжечкового червя, через который 4 желудочек сообщается с кистой ЗЧЯ. Эти изменения можно выявить при УЗ и КТ/ЯМРТ мозга как в постнатальном, так и антенатальном периоде.
При УЗИ особое внимание нижней части мозжечка, хотя для окончательного диагноза все же необходимо проведение ЯМРТ мозга. Но при ЯМРТ возможны трудности при интерпретации результатов, поэтому идет поиск наиболее информативных методов диагностики- некоторые авторы предлагают радиоизотопные методы исследования, другие – использовать пневмоэнцефалографию. Обязательно кариотипирование для исключения хромосомной патологии. Основное проявление – прогрессирующая гидроцефалия при закрытой форме синдрома СДУ.




Источник
► Синдром Денди-Уокера – это порок развития головного мозга, для которого характерна триада признаков:
■ гипоплазия червя (и полушарий) мозжечка;
■ расширение III желудочка до формирования кисты задней черепной ямки из-за ее увеличения, связанного со смещением вверх латеральных синусов и мозжечкового намета);
■ внутренняя гидроцефалия.
Специалисты пренатальной функциональной диагностики выделяют полную и неполную, а также закрытую и открытую формы синдрома Денди-Уокера. Полная форма характеризуется агенезией червя мозжечка и наличием явной коммуникации между IV желудочком и кистой в области большой цистерны. Неполная форма – это частичная агенезия нижней части червя мозжечка, в связи с чем коммуникация IV желудочка с кистой большой цистерны прослеживается не на всем протяжении червя. Открытая и закрытая формы различаются наличием или отсутствием окклюзии отверстий Люшке и Мажанди и сообщением желудочка с подпаутинным пространством.[анатомическая схема синдрома Денди-Уокера]
В 1989 г. A.J. Barkovich и соавт. предложили классификацию ликворных скоплений в задней черепной ямке, основанную на данных магнитно-резонансной томографии (МРТ). Согласно этой классификации, они описывают понятие «комплекс Денди-Уокера», которое включает четыре разновидности аномалии:
АДУ (классическая) – характеризуется увеличением задней черепной ямки с кистозной дилатацией IV желудочка; для нее типично высокое расположение намета мозжечка; поперечный синус находится выше ламбдовидного шва; отмечаются частичная или полная агенезия червя мозжечка, гипоплазия полушарий мозжечка; ствол мозга смещен к скату, большая цистерна не прослеживается, гидроцефалия присутствует в 80% случаев; при данной патологии нет сообщения расширенного IV желудочка с субарахноидальным пространством; этот тип аномалии часто ассоциируется с агенезией мозолистого тела и энцефалоцеле; клиническая манифестация этой патологии происходит практически в момент рождения;
вариант Денди-Уокера – характерны менее грубые структурные изменения: гипоплазия нижней части червя мозжечка и сообщение IV желудочка с большой затылочной цистерной через перфорированную мембрану, закрывающую выход из IV желудочка; расположение намета мозжечка и размер задней черепной ямки не изменены; ствол головного мозга не сдавлен, гидроцефалия встречается редко;
киста кармана Блейка – отличительные признаки кисты кармана Блейка: тетравентрикулярная гидроцефалия, инфра- или ретроцеребеллярная локализация кисты, сравнительно хорошо развитый червь мозжечка, кистозное расширение IV желудочка без сообщения его с большой затылочной цистерной; в эмбриогенезе карман Блейка представляет собой переходный пальцеобразный выступ заднего мембранного пространства крыши IV желудочка;
mega cisterna magna (фокальное увеличение субарахноидального пространства в нижних и задних отделах задней черепной ямки) – наблюдается увеличение размера большой затылочной цистерны; при этом цистерна свободно сообщается с IV желудочком и со смежными отделами субарахноидального пространства; увеличенная цистерна доходит до прямого синуса сверху и
до уровня СI-СII снизу.
Церебральные аномалии, связанные с синдромом Денди-Уокера, могут включать явления дисплазии (нейрональная гетеротопия, шизэнцефалия, дисгенезия мозолистого тела, голопрозэнцефалия и стеноз сильвиева водопровода; иногда может полностью отсутствовать червь мозжечка). Теперь эти находки принято расценивать как вариант синдрома Денди-Уокера.
Согласно современным представлениям, этиология синдрома Денди-Уокера чрезвычайно гетерогенна, так как в его возникновении принимают участие разные факторы: наследственные (хромосомные и генные) и экзогенные – тератогены (предполагается, что определенную роль играют и такие факторы как вирусная инфекция [ЦМВ, краснуха], алкоголь, диабет беременной). В 1/3 – 1/2 случаев синдром Денди-Уокера сочетается с различными врожденными синдромами (см. таблицу).
Среди основных гипотез возникновения синдрома Денди-Уокера можно выделить следующие: остановка эмбрионального развития в процессе формирования ромбовидного мозга, атрезия выходного отверстия из IV желудочка при отсроченном открытии отверстия Мажанди, возникновение сосудистого сплетения IV желудочка в середине тонкой крыши ромбовидного мозга.
Синдром Денди-Уокера характеризуется выраженным клиническим полиморфизмом: от практически нормального постнатального развития (имеются данные о том, что нормальное когнитивное развитие зарегистрировано у 50 % детей с синдромом Денди-Уокера; описаны случаи выявления синдрома Денди-Уокера у взрослых при плановом обследовании) до тяжелой инвалидности и даже гибели ребенка (как правило, постнатальное развитие детей имеет более серьезное отклонение в неврологическом статусе). Перинатальные исходы во многом зависят от глубины поражения ЦНС (нарастающая гидроцефалия), а также от наличия сочетанной патологии, которая в 60 – 75% случаев сопровождает данный синдром. По данным литературы, показатели постнатальной заболеваемости и смертности выше в тех ситуациях, когда синдром диагностирован в пренатальном периоде.
В случае серьезных отклонений в неврологическом статусе при наличии у ребенка Денди-Уокера в клинической картине на первый план выступают симптомы гидроцефалии в виде увеличения окружности головы, выбухания большого родничка. У новорожденного отмечается повышенная возбудимость (в т.ч. гиперрефлексия), выражена глазная симптоматика (спонтанный нистагм, косоглазие), эпизоды апноэ, парез лицевого нерва. Однако в этот ранний период жизни выявить мозжечковые симптомы не удается. Неврологические дисфункции у обычно обусловлены компрессией мозговых структур. Череп новорожденных увеличивается в размерах быстрее в окципитальной, чем во фронтальной области. Также для детей с синдромом Денди-Уокера характерен быстрый рост окружности головы в течение первых 2 месяцев жизни и расхождение черепных швов больше сзади или спереди (в такой же последовательности происходит истончение костей черепа).
Несмотря на грубый дефицит ткани червя и полушарий мозжечка, выраженные признаки атаксии совсем не являются облигатными для аномалии Денди-Уокера (наблюдаются лишь в трети случаев). Чаще выявляется недостаточность высших психических функций, к которым, в частности, относятся не только известные когнитивные нарушения, но и выявляемая у больных общая двигательная неловкость. Наибольший интерес привлекает сочетание гипоплазии червя мозжечка с различными аутистическими проявлениями, которые наблюдаются примерно в 25% случаев аномалии Денди-Уокера. В литературе последних 15 – 20 лет делаются попытки связать детский аутизм напрямую с патологией самого мозжечка и его связей с ассоциативными зонами коры большого мозга.
Изменения, характерные для синдрома Денди-Уокера можно выявить при нейросонографии (УЗИ головного мозга) в рамках пренатального скрининга. Несмотря на совершенствование пренатальной УЗ-диагностики, которая позволяет выявлять синдром Денди-Уокера уже с 15-16 недель гестации (согласно данным некоторых авторов, наиболее ранняя пренатальная диагностика аномалии Денди-Уокера была осуществлена при трансвагинальной эхографии в 12 – 14 недель беременности), в ряде случаев авторы отмечают объективные сложности визуализации тонких структур головного мозга в ранние сроки беременности. В частности, размер IV желудочка до 22 недель беременности не превышает 1 мм (!) и лишь к сроку доношенной беременности увеличивается до 3 – 4 мм.
Для своевременного установления диагноза «синдром (аномалия) Денди-Уокера» рекомендуется следующий план обследования ребенка:
■ проведение магнитно-резонансной томографии головного мозга с сагиттальным и аксиллярным обзором IV желудочка;
■ поиск других аномалий развития головного мозга;
■ офтальмологическое обследование;
■ эхокардиография;
■ кариотипирование при наличии других пороков развития;
■ консультация нейрохирурга.
Лечение. В случае отсутствия гидроцефалии и признаков внутричерепной гипертензии – наблюдение педиатра и невролога (! при необходимости – нейрохирурга). При наличии симптомов внутричерепной гипертензии – шунтирующие операции. С 1983 г. проводятся попытки внутриутробного шунтирования (постановка вентрикуло-амниотического шунта).
читайте также:
статья «Клинический случай наследственной гидроцефалии (синдром Денди – Уокера)» Ю.В. Черненков – ФГБОУ ВО «Саратовский ГМУ им. В.И. Разумовского» Минздрава России, проректор по науке, заведующий кафедрой госпитальной педиатрии и неонатологии, профессор, доктор медицинских наук; В.Н. Нечаев – ФГБОУ ВО «Саратовский ГМУ им. В.И. Разумовского» Минздрава России, доцент кафедры госпитальной педиатрии и неонатологии, кандидат медицинских наук; Ю.В. Лысова – ФГБОУ ВО «Саратовский ГМУ им. В.И. Разумовского» Минздрава России, ординатор кафедры госпитальной педиатрии и неонатологии (Саратовский научно-медицинский журнал. 2016. Т. 12, № 4) страницы: [1] [2] [3] [4];
статья «Аномалия Денди-Уокера – редкая причина сирингомиелии у взрослых» Евзиков Г.Ю., Башлачев М.Г., Белозерских К.А., Парфенов В.А.; Кафедра нервных болезней и нейрохирургии ФГАОУ ВО «Первый Московский государственный медицинский университет им. И.М. Сеченова» Минздрава России, Москва (журнал «Неврология, нейропсихиатрия, психосоматика» №9(3), 2017) [читать]
Источник