
Синдром джервелла ланге нильсена лечение

Синдром Жервелла и Ланге-Нильсена. Кардиоаудиторный или сурдокардиальный синдромJervell и Lange-Nielsen описали синдром глубокой врожденной нейросенсорной глухоты с патологией ЭКГ, характеризующейся удлинением интервала Q—Т, обморочными приступами и иногда внезапной необъяснимой смертью в детском возрасте. Впоследствии синдром был изучен многими авторами (Lcvine, Woodworth, Fraser, Froggatt, James, Jervell, Thingstad, Endsjo, Lisker, Finkelstein, Kallfelz, Sanchez Cascos et al., van Bruggen et al., Fauchier et al., Athanasiou, Weiner). Fraser, Froggatt и Murphy установили его частоту, составляющую от 3 до 4 случаев на 1 млн. рождений. Кроме того, они определили, что синдром может составлять до 1% среди наследственной глухоты. Однако Sanchez Cascos с сотр. нашли только один случай среди 511 глухонемых детей. Fay с соавт. обнаружили одного больного с этим синдромом среди 1100 глухих детей. Кроме того, 4 больных страдали обмороками без удлинения интервала Q—Т, а у 5 больных отмечалось удлинение интервала Q—Т без обмороков. Орган слуха. У всех больных отмечалась врожденная двусторонняя глубокая нейросенсорная глухота. Лабораторные данные. Изменения ЭКГ характеризовались удлинением интервала Q—Т с широкой T-волной, которая могла быть вертикальной, зазубренной, двухфазной или перевернутой. Степень удлинения интервала Q—Т варьировала как внутри одной семьи, так и между разными семьями, но почти всегда превышала 0,5 с (при максимуме 0,4 с В норме). Так как интервал Q—Т удлиняется в зависимости от скорости сердечных сокращений, существует простая формула для определения его величины (Ljting): Q—T= (RRX0,2) +0,18±0,04. В нескольких случаях была обнаружена легкая гипохромная анемия (Jervell, Langc-Nielsen, Fraser, Froggatt, James, Jervell et al., Lamy et al, Kallfelz). Патология. Описаны многочисленные результаты вскрытия. При макроскопическом и гистологическом исследовании сердца в большей части случаев патологии не обнаружили. Специальное исследование проводящей системы сердца показало выраженное сужение главной ветви артерии синусового узла, в результате чего развивался инфаркт узла (Friedmann et al.). Fraser, Froggatt и James обнаружили, что в сердце этих больных отсутствует нормальная гликогенсодержащая перинуклеарная зона в волокнах Пуркинье.
Изменения височной кости описаны Friedmann с соавт.. Наиболее уникальным изменением являлось накопление ПАСК-положительных глыбок гиалина в атрофичной сосудистой полоске. Отмечалась почти полная дегенерация кортиева органа с утратой чувствительных клеток. Покровная мембрана была сморщенной или втянутой, а рейснерова мембрана была сращена с основной мембраной, практически облитсрируя улитковый ход. Чувствительный эпителий утрикулюса и саккулюса был атрофичным, а гребешок дезорганизованным. Отмечалась умеренная утрата нервных клеток спирального узла. Наследственность. Синдром отчетливо наследуется по аутосомно-рецессивному типу. Часто встречаются кровнородственные браки между родителями (Fraser, Froggatt, Murphy, Lamy et al., Sanchez Cascos et al.). У гетерозигот может наблюдаться умеренное удлинение интервала Q—Т (Fraser, Froggatt и Murphy). Предполагают, что заболевание наследуется сцепленно с Rh-фактором (Friedmann, Fraser, Froggatt). Диагноз. Описаны некоторые изменения ЭКГ (такие, как удлинение интервала Q—Т) без глухоты (Johansson, Jorming, van der Straaten, Bruins, Singer et al.). В этих случаях также наблюдались внезапная смерть и/или приступы потери сознания. Однако патология в этих семьях наследовалась по доминантному типу. Jervell описал еще один доминантно наследующийся синдром множественных экстрасистол с приступами фибрилляции желудочков, но с нормальным интервалом Q—Т. Отмечается определенное сходство настоящего заболевания с изменениями, наблюдающимися при синдроме Рефсума. При обоих синдромах выражены нейросенсорная глухота, нарушения сердечной проводимости с удлинением интервала Q—Т и аномальной Т-волной и иногда наступает внезапная смерть. Однако при синдроме Рефсума глухота выявляется в зрелом возрасте и, кроме того, в сыворотке крови повышен уровень фитановой кислоты. Обморочные приступы при синдроме Ланге-Нильсена могут быть ошибочно диагностированы как эпилептические припадки. Однако ЭЭГ нормальна, в то время как ЭКГ резко изменена. Кроме того, после обморочных состояний у детей не развивается глубокого оглушения. Отмечено несколько атипичных случаев. Mathews с соавт. сообщили об аутосомно-доминантной форме с легкой высокочастотной глухотой. Глухота и сердечная патология могут наследоваться как независимые доминантные признаки. Furlancllo с сотр. и Athanasiou и Muller-Seydlitz также описали доминантную форму заболевания у взрослых с легким дефектом слуха. Предполагают также, что в этих семьях имелся синдром «LEOPARD». Читатель, конечно, помнит, что причиной удлинения интервала Q—Т могут служить: гипокалиемия, гипокальциемия, гипомагниемия и введение квинидина и фенотиазина. Прогноз. С возрастом отмечается незначительное прогрессирование дефекта слуха. У больных может наблюдаться различное число обморочных приступов. Примерно в половине случаев больные погибают к 15-летнему возрасту. Было выявлено несколько больных в возрасте старше 21 года. Выводы. Главными чертами этого синдрома являются: – Также рекомендуем “Ушная-зубная или отодентальная дисплазия. Липодистрофия лица, плеч и кисты костей с глухотой” Оглавление темы “Генетические болезни с глухотой”:
|
Источник
Медицинская статья опубликована в рубрике: Кардиология, ЛОР, СИНДРОМЫ | Декабрь 9th, 2013
В 1957 г. Жервелл совместно с Ланге Нильсеном описали синдром, характеризующийся постоянным сочетанием врожденной глухоты, с приступами синкопы и характерными электрокардио графическими изменениями.
Синдром Жервелла Ланге Нильсена — генетическая аномалия с семейным характером, поражающая оба пола.
Этиопатогенез синдрома Жервелла Ланге Нильсена.
Не существует предположения, которое объяснило бы одновременное нарушение слуха и миокарда. Предположения тех, кто придерживается единого мнения, объясняет аномалию сердца гипокалиемией, а глухоту — нарушением кровоснабжения всего слухового аппарата.
Удлинение интервала Q — Т обусловлено нарушением реполяризации (с неуточненной этиологией), с переменчивой интенсивностью от одного дня к другому, определяя такое же переменчивое изменение интервала Q — Т.
Сердечное синкопе объясняется, как приступ типа Адамса-Стокса, или как приступ венечной ишемии. Смерть, наступившая при этих синкопах, может быть вызвана бесконечным удлинением интервала Q—Т, полной предсердно-желудочковой диссоциацией или фибрилляцией желудочков. Следует подчеркнуть, что синкопе появляются все реже и реже, по мере развития ребенка.
Синдром встречается, как у взрослых, так и у детей; по Fraser, представляет приблизительно 1% всех случаев врожденной глухонемоты. Наследственная передача происходит рецессивным образом, вероятно, как результат палеотропического эффекта аномального гена у гомозигот с полным клиническим проявлением; у гетерозигот синдром проявляется только удлинением интервала Q—T.
Симптоматология синдрома Жервелла Ланге Нильсена.
Врожденная глухота — всегда присутствует; она двусторонняя, обусловливая глухонемоту маленького ребенка. Глухота перцепторного типа, не затрагивает низкие звуки.
Приступы синкопе — повторные, вызываемые обыкновенно волнениями, холодом или даже без видимой причины, продолжаются несколько секунд и внезапно прекращаются.
Во время синкопы у больных отмечается:
- бледность;
- тахикардия;
- полипное;
- больной стонет, теряет или нет сознание;
- иногда появляются судороги.
Несколько минут после прекращения синкопе, у больного затуманенное сознание, затем постепенно приходит в нормальное состояние.
Диагностика синдрома Жервелла Ланге Нильсена.
Электрокардиографические характерные изменения заключаются в:
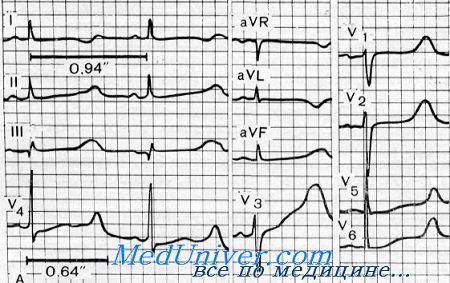
— патологическом удлинении интервала Q—Т (представляя постоянные и значительные электрокардиографические изменения). В сравнении с нормальным пределом этого интервала (0,31 секунды) величины, встречающиеся у больных, всегда намного выше (в среднем они колеблются между 0,40 и 0,55 секунд при сердечном ритме выше 90/мин).
Фонокардиограмма показывает, что длительность электрической систолы всегда больше механической:
- изменения волны Т очень разнообразна: отрицательная волна, положительная и двухфазная;
- комплекс QRS и интервал Р — Q — всегда нормальные.
Непостоянно добавляется и гипохромная анемия, умеренной интенсивности.
Течение и прогноз синдрома Жервелла Ланге Нильсена.
Течение, как и дальнейший прогноз, неблагоприятные. Смерть ребенка может произойти во время приступа синкопе, тем более, что часто приступ синкопе диагностируется, как приступ эпилепсии и лечат его, как таковой, лишая, таким образом, больного соответствующей экстренной терапии.
Лечение синдрома Жервелла Ланге Нильсена
а) Лечебное:
- экстренное назначение (если диагноз был уточнен) дигиталиса в 1% растворе, 5—6 капель в день, 5 дней в неделю (с еженедельным 2-х дневным перерывом во избежание накопления дигиталиса в миокарде), в течение 3—4 недель. Применением дигиталиса можно избежать смерти во время синкоп, укорачивая и нормализируя интервал Q — R, в отличие от хинидина, который способствует его удлинению (отсюда вытекает абсолютное противопоказание хинидина при приступах синкопе);
- хлористый калий в дозе от 0,5—1 г/день перорально;
- успокаивающие (диазепам).
б) Профилактическое:
- электрокардиографическое исследование всех новорожденных и грудных детей, страдающих судорогами и состоянием синкопе;
- внимание при дифференциальном диагнозе приступа синкопе, который ошибочно может быть принят за приступ эпилепсии; от своевременного и правильного диагноза зависит жизнь больного.
Источник
Год утверждения 2016
Профессиональные ассоциации:
- Ассоциация детских кардиологов России
- Союз педиатров России
Оглавление
1. Краткая информация
2. Диагностика
3. Лечение
4. Реабилитация
5. Профилактика
6. Дополнительная информация
1. Краткая информация
1.1 Определение
Синдром удлиненного интервала QT (СУИQT) – наследственное заболевание с высоким риском ВСС, характеризующееся увеличением QT, приступами потери сознания на фоне желудочковых аритмий.
1.2 Этиология и патогенез
Синдром обусловлен мутациями в генах, кодирующих альфа- и бета-субъединицы ионных каналов мембраны кардиомиоцита и белки, участвующие в регуляции ионных токов.
Мутации в генах выявляют в 50-75% случаев.
Нарушение функции ион-специфических каналов приводит к изменению скорости ионных токов и увеличению продолжительности потенциала действия.
Врожденные мутации приводят к полной или частичной потере функции пораженного канала.
Удлинение QT – триггерный фактор для жизнеугрожающей полиморфной желудочковой тахикардии типа «пируэт».
Тахикардия типа «пируэт» – частая причина внезапной смерти при СУИQT.
К удлинению интервала QT также приводит симпатический дисбаланс при левосторонней симпатической иннервации сердца.
1.3 Эпидемиология
Распространенность СУИQT – один случай на 2500-3000 новорожденных.
Среди генотипированных больных «немые» мутации – у 10-36%.
1.4 Кодирование по МКБ-10
I 45.8 – Синдром удлиненного интервала QT
Примеры диагнозов:
- Синдром удлиненного интервала QT, первичный, бессинкопальная форма, I молекулярно-генетический вариант.
- Синдром Джервелла-Ланге-Нильсена (синдром удлиненного интервала QT, первичный, синкопальная форма, врожденная нейросенсорная тугоухость III-IV степени).
- Синдром удлиненного интервала QT, первичный, синкопальная форма, II молекулярно-генетический вариант.
1.5 Классификация
Классификация СУИQT включает 15 молекулярно-генетических вариантов.
Клиническая классификация:
- синкопе в сочетании с удлинением интервала QT;
- удлинение интервала QT в отсутствие синкопе;
- синкопе в отсутствие удлинения интервала QT;
- скрытая форма (“form frust”).
2. Диагностика
2.1 Жалобы и анамнез
Спектр клинических проявлений – от полного отсутствия симптомов до синкопальных состояний и внезапной смерти.
Патогномоничны для СУИQT синкопальные состояния, провоцированные физической и/или эмоциональной нагрузкой, резким звуком, плаванием.
Жалобы на сердцебиение перед потерей сознание редки.
Наличие у родственников 1 и 2 степени приступов потери сознания и/или случаев внезапной смерти до 40 лет; удлинение интервала QT на ЭКГ членов семьи и/или с СУИQT у родственника.
2.2 Физикальное обследование
Фенотипические особенности:
- синдром Романо-Уорда
- синдактилия в 100% синдрома Тимоти
- врожденная нейросенсорная тугоухость при синдроме Джервелла-Ланге-Нильсена
- низкий рост и сколиоз, низко посаженные уши, гипертелоризм, дефекты нёба, микрогнатия, клинодактилия, синдактилия при синдроме Андерсена-Тавила.
Синкопальные состояния манифестируют в любом возрасте, неблагоприятный прогноз – первый синкопе до 6 лет.
У мужчин риск первого синкопе выше в детском возрасте и снижается после подросткового периода.
У женщин наибольшая вероятность первого синкопе – в послеродовом периоде.
Геноспецифические провоцирующие синкопе факторы:
- плавание или ныряния при I молекулярно-генетическом варианте;
- резкий звук при II варианте;
- синкопе во сне при III варианте.
У одного пациента могут быть два и более провоцирующих фактора.
Потеря сознания около 1-2 минут, редко до 20 минут.
Иногда обморок сопровождается судорогами и непроизвольным мочеиспусканием.
Синдром Джервелла-Ланге-Нильсена:
- очень тяжёлая форма СУИQT;
- мутация в генах KCNQ1 и KCNE1;
- аутосомно-рецессивный тип наследования;
- манифестация у 15% до 1 года, у 50% – до 3 лет;
- провоцирующий фактор – нагрузка физическая/эмоциональная;
- бета-блокаторы неэффективны в большинстве случаев;
- показана имплантация кардиовертера-дефибриллятора.
Синдром Андерсена-Тавила:
- периодический калийчувствительный паралич в 100%;
- краниофасциальный и скелетный дисморфизм (сколиоз, микрогнатия и т.д.);
- мутации в гене KCNJ2;
- аутосомно-доминантный тип наследования;
- манифестирует до 10 лет или в подростковом возрасте;
Синдром Тимоти:
- полиорганные поражения;
- синдактилия;
- врожденные пороки сердца;
- иммунодефицитные состояния;
- транзиторная гипогликемия;
- когнитивные нарушения и аутизм;
- мутации в гене CACNA1С;
- наследуется по аутосомно-доминантному типу;
- средняя продолжительность жизни около 2,5 лет.
2.3 Лабораторная диагностика
Биохимический анализ крови:
- электролиты;
- активность кардиоспецифических ферментов;
- маркёры воспаления;
- титр антител к структурам сердца.
Оценка гормонального профиля щитовидной железы.
Молекулярно-генетическая верификация.
2.4 Инструментальная диагностика
ЭКГ
Критерии P. Schwartz (1993г.), усовершенствованные в 2011 году.
Поверхностная ЭКГ в 12 отведениях в клиноположении, ортоположении и после 10 приседаний со скоростью 50 мм/с.
Коррекция интервал QT по отношению к ЧСС по формуле Базетта.
Удлинение интервала QT:
для женщин >460 мс
для мужчин >450 мс.
Дисперсия интервала QT:
у здоровых взрослых от 48±18 до 54±27 мс;
у здоровых детей от 7 до 16 лет 21±11 мс.
Морфология комплекса QRST:
I вариант – зубец Т с широким основанием и косо восходящей элевацией сегмента ST;
II вариант – двугорбый/зазубренный Т в правых грудных отведениях;
III вариант – удлинение QT за счёт сегмента ST, зубец Т обычной морфологии.
Для СУИQT характерна макроальтернация зубца T – изменение полярности и амплитуды в последовательных кардиоциклах.
Альтернация зубца Т ассоциируется с желудочковой тахикардией типа «пируэт» и выраженным более 500 мс удлинением интервала QTс.
Для синдрома Тимоти характерны:
- удлинение QTc до 700 мс;
- развитие функциональной AV-блокады с проведением 2:1;
- макроальтернация зубца Т.
Для синдрома Андерсена-Тавила характерны:
- высокоамплитудные зубцы U;
- Т с покатым, растянутым нисходящим коленом.
Суточное мониторирование ЭКГ (СМЭКГ)
Для выявления маркёров электрической нестабильности миокарда, сопутствующих нарушений ритма и проводимости.
Предпочтительны системы с опцией автоматического анализа интервала QT.
При ХМ-ЭКГ дополнительно оценивают:
- продолжительность интервала QT в автоматическом режиме;
- мануальную оценку интервала QT и QTc на минимальной и максимальной ЧСС;
- особенности морфологии зубца Т;
- наличие зубца U;
- макроальтернации зубца Т.
Эхокардиография (ЭхоКГ)
- Всем больным при первичном обследовании для исключения органической патологии сердца, оценки электромеханического соответствия систолы желудочков на фоне изменения интервала QT.
- Измеряется соотношение времени электрической и механической систолы желудочков.
Тест с физической нагрузкой
- Для дифференциальной диагностики.
- Для определения эффективности антиаритмической терапии.
- Продолжительность интервалов QT и QTc оценивается в исходе, на максимуме нагрузки и на восстановлении.
2.5 Иная диагностика
Дифференциальный диагноз между первичным и вторичным СУИQT, молекулярно-генетическими вариантами синдрома.
3. Лечение
Терапия больных с СУИQT:
- коррекция образа жизни с исключением препаратов, удлиняющих QT;
- медикаментозная и немедикаментозная профилактика ВСС;
- неотложная терапия желудочковой тахикардии типа «пируэт».
Профессиональный спорт противопоказан при синкопальной форме СУИQT и больным из группы высокого риска.
При отсутствии клинических проявлений и генетически подтвержденном СУИQT решение о запрете спорта принимается врачебной комиссией.
3.1 Консервативное лечение
Пожизненная антиаритмическая терапия бета-адреноблокаторами с коррекцией дозы по мере роста пациента.
Бета-адреноблокаторы показаны:
- бессимптомным пациентам с QTc >470 мс;
- пациентам с синкопой;
- при документированной желудочковой тахикардии/фибрилляции желудочков.
Применяются бета-адреноблокаторы:
- неселективный пропранолол0-4.0 мг/кг/сут в 3-4 приёма;
- неселективный нодалол5-1.0 мг/кг/сут в 1-2 приёма;
- селективный атенолол5-2.0 мг/кг/сут в 2 приёма.
Не рекомендуется метопролол, повышающий риск рецидива синкопе.
При III варианте СУИQT и QTc >500 мс с уменьшением QTc более 40 мс после лекарственной пробы вместе с бета-адреноблокатором назначается блокатор натриевых каналов – мексилетин 2.0-5.0 мг/кг на 3 приёма.
3.2 Хирургическое лечение
Имплантация кардиовертера-дефибриллятора для профилактики ВСС показана всем больным:
- перенесшим ВОК;
- при наличии спонтанной устойчивой желудочковой тахикардии с/без синкопе;
- рецидивирующих на фоне антиаритмической терапии синкопе.
Левостороння симпатэктомия рекомендована:
- при сохранении рецидивов желудочковой тахикардии на фоне максимально допустимой дозы бета-блокаторов;
- при противопоказаниях или непереносимости бета-блокаторов;
- при противопоказаниях к имплантации ИКД;
- при отказе от имплантации ИКД.
4. Реабилитация
Медицинской и физической реабилитации больных не требуется.
Детям с частыми срабатываниями ИКД – консультация психолога.
Показано санаторно-курортное лечение в санаториях кардиологического профиля.
5. Профилактика и диспансерное наблюдение
5.1. Профилактика
Для профилактики рецидива желудочковой тахикардии и ВСС необходим динамический контроль факторов риска.
Для своевременной диагностики заболевания обследуют группы риска – членов семьи:
- I и II степени родства больного СУИQT;
- внезапно умерших в молодом возрасте.
5.2. Ведение пациентов
Больные с генетически детерминированными нарушениями ритма наблюдаются в специализированном аритмологическом центре.
Частота посещений зависит от возраста больного и тяжести заболевания.
Контроль эффективности терапии и мониторинг факторов риска:
- при синкопальной форме – 1 раз в 1-6 месяцев;
- без синкопе – не реже 1 раза в год;
- в пубертатном периоде – 1 раз в 6 месяцев.
Первичная госпитализация в специализированное кардиологическое отделение для диагностики и стратификации индивидуального риска ВСС.
Продолжительность госпитализации определяется основным заболеванием.
Контроль системы ИКД у пациентов с имплантированным кардиовертером-дефибриллятором:
- не реже 1 раза в 6 месяцев;
- каждый раз при срабатывании устройства;
- при рецидиве синкопе.
При плановом контроле системы ИКД предварительно выполняют:
- ЭКГ;
- ХМ;
- ЭхоКГ;
- рентгенографию органов грудной клетки в прямой и левой боковой проекциях.
Вакцинация
Решение о вакцинации индивидуальное в зависимости от:
- состояния пациента;
- эффективности медикаментозного контроля аритмии;
- с учётом ранее выявленных провоцирующих факторов.
Детям с синкопальной формой СУИQT вакцинация по индивидуальному графику.
В отсутствие синкопе вакцинация проводится в декретированные сроки.
6. Дополнительная информация, влияющая на течение и исход заболевания
Исходы и прогноз
Прогноз заболевания основывается на риске ВС и зависит от молекулярно-генетического варианта синдрома, возраста манифестации синкопальных состояний, эффективности антиаритмической терапии бета-блокаторами.
Прогноз для жизни благоприятный при регулярном мониторинге факторов риска ВС и своевременной коррекции модифицируемых факторов риска.
Источник