
Синдром герстманна штреусслера шейнкера лечение

1.Общие сведения
Синдром (болезнь) Герстмана-Штраусслера-Шейнкера, или синдром ГШШ – нейродегенеративное прионное заболевание. За этим исчерпывающим и сугубо специальным определением скрывается тяжелая, редкая, в некоторых отношениях уникальная патология, и хотя бы поэтому каждое слово заслуживает более подробного рассмотрения.
Дегенерацией, – применительно к единичному организму, – называют патологический процесс перерождения (вырождения) на клеточном уровне, в ходе которого та или иная ткань постепенно утрачивает нормальную, естественную для нее структуру, становясь все более примитивной по строению и все хуже справляясь с возложенными на нее функциями. Как правило, такие процессы обусловлены нарушениями клеточного метаболизма (дистрофия) и/или дефицитом кровоснабжения (ишемия), конечным результатом чего является полная деградация, сокращение в объеме и отмирание (атрофия, некроз, инфаркт) паренхиматозной, т.е. функциональной, узкоспециализированной ткани какого-либо органа. Соответственно, термином «нейродегенеративные заболевания» обозначают большую группу болезней, обычно наследственных и на сегодняшний день неизлечимых, при которых дегенерирует сложнейшая паренхима центральной нервной системы, т.е. нейронное вещество спинного и/или головного мозга, а также нервных узлов и сплетений периферической нервной системы. Начинаясь, как правило, с незначительных нарушений памяти, речи, моторных функций, большинство таких заболеваний в терминальной стадии обусловливают тотальную деменцию (органическое слабоумие) и сугубо вегетативное существование пациента. К наиболее известным нейродегенеративным процессам относятся болезни Альцгеймера, Пика, Паркинсона, амиотрофический боковой и рассеянный склероз, и пр.
Прионные же заболевания, или спонгиоформные (губчатые) энцефалопатии – совершенно особый вид нейродегенерации, причиной которого является недавно открытые (1982) белки-прионы, до сих пор остающиеся предметом интенсивных научных дискуссий: некоторые исследователи склонны считать прионы особой формой жизни, другие рассматривают их исключительно как биохимический феномен. Так или иначе, но прионные молекулы способны in vivo модифицировать другие белки таким образом, что те превращаются в идентичные прионы; в результате клетка погибает, а ткань в целом вырождается в пористую биомассу, не имеющую никакого отношения к первоначальной специализации. Поскольку этот процесс прогрессирует в веществе головного мозга (причем значительно быстрее, чем другие виды нейродегенерации), его проявления и последствия трудно назвать иначе, как катастрофическими. Широкую огласку получили прионные эпизоотии сельскохозяйственного скота (овечья почесуха, коровье бешенство), однако прионными оказались и некоторые заболевания человека, ранее приписываемые неизвестным вирусам, – болезнь Крейтцфельдта-Якоба, куру, фатальная семейная инсомния.
Что касается синдрома Герстмана-Штраусслера-Шейнкера, то как самостоятельная нозологическая единица он выделен и описан еще в 1936 году, однако этиопатогенез этой специфической нейродегенерации стал ясен лишь с открытием прионов и осуществлением хромосомных исследований. К счастью, эта патология не просто редка, а встречается исключительно редко: примерно в одном случае на 10 млн населения.
2.Причины
Наряду с фатальной семейной бессонницей и генетическим вариантом болезни Крейтцфельдта-Якоба, синдром ГШШ является не трансмиссивным (заразным), а наследственным заболеванием. Дефектный ген PRPN запускает «цепную реакцию» воспроизводства белков-прионов в мозговой ткани. Как правило, это происходит в возрастном интервале от 20 до 60 лет, однако о реальной распространенности, о возрастных закономерностях, о влиянии фактора пола и других характеристиках процесса трудно судить с уверенностью, поскольку накопленный объем наблюдений, по статистическим меркам, совершенно недостаточен.
3.Симптоматика, диагностика
Некоторые клинические и патоморфологические особенности синдрома ГШШ роднят его с болезнью Альцгеймера, другие – с различными нейродегенеративными формами атаксии (неупорядоченности, двигательной дискоординации). Это дополнительно осложняет диагностику и, видимо, нередко приводит к диагностическим ошибкам, которые могут быть однозначно устранены только при посмертном патоморфологическом исследовании.
В симптоматике выявляются феномены, характерные как для органического поражения мозжечка (нарушения координации движения, способности к пространственной ориентации), так и т.н. бульбарная симптоматика, специфическая для поражений продолговатого мозга: ослабление и постепенное отмирание глотательного рефлекса, прогрессирующие нарушения артикуляторных составляющих речи и т.д. В терминальной стадии клиническая картина носит столь же тотальный и катастрофический характер, как и при других видах нейродегенерации. Однако от сходных заболеваний именно прионного генеза синдром ГШШ отличается более медленным течением: усредненный период от манифестации до летального исхода составляет примерно 5-6 лет (в разбросе от 3 месяцев до 13 лет).
4.Лечение
Как отмечалось выше, этиопатогенетического лечения (устраняющего причину либо хотя бы влияющего на механизмы развития) прионных и/или нейродегенеративных заболеваний на сегодняшний день не существует. Проводится симптоматическое лечение (ноотропы, протекторы когнитивных функций, корректоры поведения и пр.). Однако одна из уникальных особенностей синдрома ГШШ заключается в том, что в 2014 году было опубликовано сообщение о том, что путем сложнейшей генетической профилактики и последующего экстракорпорального оплодотворения удалось предотвратить «запуск» прионной нейродегенерации у детей пациентки с очень высоким риском хромосомной передачи именно этого заболевания. Разумеется, от одной публикации до всесторонне аргументированной, доказанной и отработанной клинической методики – дистанция огромная, и все же есть, по всей вероятности, реальные основания предполагать решение проблемы прионных нейродегенеративных заболеваний в обозримом будущем.
Источник
Неврология
10.11.2018
 Одно из тяжелых неврологических расстройств головного мозга приводит к тому, что страдающий им человек перестает ощущать собственное тело.
Одно из тяжелых неврологических расстройств головного мозга приводит к тому, что страдающий им человек перестает ощущать собственное тело.
Хотя синдром Герстманна-Штреусслера-Шейнкера встречается редко, он может поразить взрослого человека практически в любом возрасте: опасен промежуток от 20 до 60 лет.
Для проявления заболевания необходимо совпадение двух факторов – семейной предрасположенности и накопления патологического прионного белка.
Общие сведения о болезни
Синдром Герстманна относится к медленным вирусным инфекциям. Болезнь входит в группу губкообразных энцефалопатий. Ее можно диагностировать по дегенеративным поражениям центральной нервной системы, при которых:
- формируется губкообразное состояние головного мозга;
- образуется много амилоидных бляшек в мозговых отделах;
- развивается атаксия;
- прогрессирует слабоумие.
Патологические прионы провоцирующие опасный синдром – особенные белковые молекулы, способные к размножению. Попадая в человеческий организм, они увеличивают свое количество за счет изменения нормальных прионов. По сравнению с размножением вирусов или бактерий для этого требуется больше времени, поэтому человек заболевает не сразу, а через несколько лет.
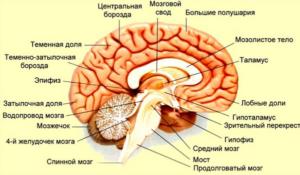 Прионы обладают выраженной устойчивостью. Их нельзя уничтожить кипячением, замораживанием, ультрафиолетовым облучением, воздействием радиации, обработкой формалином. Человеческий организм не замечает вредных клеток, не продуцирует против него антитела, не посылает в атаку лимфоциты.
Прионы обладают выраженной устойчивостью. Их нельзя уничтожить кипячением, замораживанием, ультрафиолетовым облучением, воздействием радиации, обработкой формалином. Человеческий организм не замечает вредных клеток, не продуцирует против него антитела, не посылает в атаку лимфоциты.
Больные страдают парезом взора, деменцией, гипорефлексией, глухотой, миоклоническими судорогами и патологическими подошвенными рефлексами.
Развитие болезни приводит к депрессии, вызывает приступы психоза, угрожает резкой потерей веса. Заболевание постепенно прогрессирует, оно опасно тем, что неизбежно приводит к смертельному исходу.
Причины синдрома Герстмана
Развитие болезни происходит, когда у человека поражается доминантное полушарие головного мозга. Это относится к следующим его отделам:
- оболочке;
- коре;
- долям.
К основным первопричинам относится появление опухоли любой природы (она поражает височную и теменную области мозга) или серьезный сбой в системе кровообращения. К провоцирующим факторам, которые способны подтолкнуть появление и развитие заболевания, относятся следующие:
- травма головы;
- заражение вирусной инфекцией – она угрожает серому веществу и мозговой оболочке;
- осложнения после вакцинации.
Свою негативную лепту вносят болезни, чье возникновение спровоцировала интоксикация организма, получившего серьезный удар отравляющими веществами.
Симптоматика
Заболевание Герстмана легко диагностировать, если заболевший достиг возраста 30-45 лет.
У человека с опасным недугом:
- Выпадает левая или правая половина поля зрения (это относится к двум глазам), снижается и доходит до полного отсутствия способность концентрации взгляда на предмете, находящемся в движении.
- Хотя двигательная активность рук с интеллектом сохраняется, происходит полная потеря способности к письму.
- При распространении заболевания на затылочную область исчезает понимание счета и смысла. Человек с синдромом Шейнкера не может выполнить арифметические действия, довести до логического завершения любую из операций.
- Отсутствует способность истолковать написанные слова, знаки, прочесть записи тогда, когда зрение сохранено.
- Полностью исчезает пространственная ориентация. Заболевший не знает, где расположены части его собственного тела применительно к правой или левой стороне.
Список симптомов пополняется бессонницей, двигательными расстройствами, утратой циркадных ритмов, деменцией. В самом начале заболевания следует обращать внимание на нарушения вегетативного характера: изменяются потоотделение и саливация, появляются запоры, возникают проявления тахикардии, гипертензии и тахипноэ, иногда дело доходит до лихорадок.
Лечение синдрома
Пока для облегчения состояния заболевании возможна только поддерживающая терапия. Перед назначением лечения Герстманна-Штреусслера-Шейнкера, врач выявляет какая из основополагающих причин обусловила развитие заболевания и запустила формирование механизма, губительного для структур головного мозга.
Поскольку болезненная патология изучена не полностью, эффективная патогенетическая терапия в стадии разработки. При правильно подобранных лекарственных средствах и регулярном их применении можно устранить симптомы либо предотвратить их стремительное развитие.
Результативна симптоматическая терапия с применением противосудорожных средств – они способны существенно облегчить страдания. Для лечения используют препараты:
- Ноотропного воздействия. С их помощью улучшается уровень проведения внутримозговых импульсов. Чаще всего прописывается прием Ноотропила или Котексина.
- Для улучшения мозгового кровотока. Речь идет о Винпоцетине и Пентоксифиллине.
- Помогающие скорректировать поведение заболевшего. При наличии показаний он принимает антидепрессанты с барбитуратами и транквилизаторами.
Кроме медикаментозной терапии, облегчение приносят физиотерапевтические методы – от курсов лечебной физкультуры и массажа до регулярного плавания и занятий водной гимнастикой.
Неплохо укрепляет организм санаторно-курортное лечение. Оно помогает сохранять тонус активным и поддерживать жизненные силы на приемлемом уровне.
Источник
Синдром Герстмана (синдром Герстмана-Шильдера, синдром угловой извилины) – неврологическое расстройство, представленное рядом симптомов, указывающих на наличие поражения определённой области мозга. Названо в честь австрийского невролога Йозефа Герстмана (Josef Gerstmann, 1887–1969).
Синдром Герстмана Штреусслера, в свою очередь, представляет собой семейную наследственную прионовую инфекцию, входящую в группу трансмиссивных энцефалопатий, к которым относятся болезнь Крейтцфельда-Якоба и Куру.
Появление симптомов нарушения когнитивных функций может указывать на множество серьезных патологий головного мозга, к одной из них относится синдром Герстмана. Своевременное обращение к врачу поможет предотвратить появление осложнений, благодаря правильной диагностике и лечению недуга.
Причины возникновения
Провоцирующие факторы развития данной патологии следующие:
- воспалительные заболевания (энцефалит, менингит, сепсис с распространением инфекции на структуры мозга);
- эмболический процесс (на фоне тромбообразования);
- состояние после проведенной вакцинации;
- атеросклероз (при АГ, повышенном содержании холестерина в крови);
- опухолевидные образования (кисты, опухоли, абсцессы, гематомы);
- травмы (ушиб, сотрясение);
- общее переутомление.

Характерная симптоматика
У пациента с синдромом угловой извилины можно выявить следующие специфические признаки:
- Контрлатеральная гомонимная гемианопсия (выпадение правых или левых половин полей зрения обоих глаз) в совокупности со снижением или отсутствием способности концентрировать взор на движущемся предмете (ортокинетический нистагм).
- Аграфия (потеря способности к письменной речи с сохранением координированной двигательной активности рук и интеллекта). Наблюдается при патологии задних отделов лобных извилин правого (у левши) или левого (у правши) полушарий ГМ.
- Акалькулия (невозможность выполнения счетных операций, арифметических действий, пациент не понимает счёт и его смысл). Если патология затрагивает теменно-затылочную область, то развивается этот симптом.
- Алексия (пациент не способен дать толкование написанным словам, знакам, записям при сохраненном зрении).
- Пальцевую агнозию и отсутствие пространственной ориентации (невозможность определить место нахождения и расположение собственных частей тела относительно правой или левой сторон).
На фото представлены основные вышеперечисленные проявления заболевания.
Клинические признаки патологии отмечаются в среднем возрасте (35–40 лет) и схожи с таковыми при заболевании Крейтцфельда-Якоба. Отличительными симптомами выступают отсутствие деменции и более длительный период жизни (более 5 лет) при синдроме Герстмана.
Постановка диагноза
Для подтверждения наличия у больного синдрома Герстмана Штреусслера Шейнкера необходимо провести ряд исследований:
- Определение мутации в ДНК на уровне гена PRNP. Проводится автоматическое секвенирование, с помощью которого врач исследует нуклеотидную последовательность гена, чтобы обнаружить дефект.
- МРТ головного мозга (для выявления пораженных очагов на любой стадии заболевания, изменений в области коры или полушарий мозга, ущемленных нервов или сосудов).
- Анализ крови на наличие прионовых тел (иммуно-блоттинг периферических лимфоцитов).
- Состояние ликвора (иммуноферментный анализ помогает определить нейроспецифический белок).
Тесты
Кроме генетического обследования, в постановке диагноза важная роль отводится когнитивным тестам, с их помощью можно точно определить наличие у пациента интеллектуальных расстройств. Они представлены списком несложных вопросов и заданий, помогающих в определении патологии или риска ее развития у каждого пациента. Для этого врач просит больного:
- взять ручку, или другой предмет в правую или левую руку (для определения нарушений пространственной ориентации);
- написать текст, предложение, словосочетания (для выявления аграфии);
- прочитать отрывок, предложение, текст (с целью исключения алексии);
- провести простые счетные операции на сложение или вычитание (на диагностику акалькулии);
- остановить взгляд на движущемся перед глазами предмете (способность утрачена при данной патологии);
Поддерживающая терапия
Перед назначением лечения патологического состояния врач выявляет основополагающую причину, обусловившую развитие заболевания и механизм поражения структур мозга. Такая патология, как синдром Герстмана, на сегодняшний день изучена не до конца, вследствие чего не удается разработать эффективной патогенетической терапии. Лечебная тактика направлена на устранение основных симптомов и предотвращение их прогрессирования. С этой целью используются препараты:
- ноотропного действия, способствующие улучшению проведения импульсов в мозге, («Ноотропил», «Котексин»);
- улучшающие мозговой кровоток («Пентоксифиллин», «Винпоцетин»);
- корректирующие поведение больного (антидепрессанты, транквилизаторы, барбитураты);
Активно используются методы физиотерапии: лечебная физкультура, массаж, плавание и водная гимнастика. В качестве общего укрепления организма и поддержания жизненных сил и тонуса полезным будет санаторно-курортное лечение.
Загрузка…
Источник
Синдром Герстмана – Штраусслера – Шейнкера (болезнь Герстмана – Штраусслера –
Шейнкера, синдром ГШШ, подострая губкообразная энцефалопатия, МКБ 10 – A81.9) —
чрезвычайно редкое смертельное нейродегенеративное заболевание, относящееся к группе
прионных болезней. Синдром Герстмана – Штраусслера – Шейнкера характеризуется
мозжечковыми нарушениями, параличами, экстрапирамидными нарушениями
(двигательные нарушения, возникающие в результате повреждения базальных ганглиев),
глухотой, слепотой и деменцией. Начало заболевания, как правило, приходится на третье
– четвертое десятилетия жизни. Средняя продолжительность жизни после начала болезни
составляет около пяти лет.
Синдром ГШШ, как и болезнь Крейтцфельдта – Якоба, ассоциирован с мутациями
гена PRNP, кодирующего нормальный белок PrPC, участвующий в формировании
миелиновой оболочки нервных волокон, и изоформу этого белка — прионный белок
PrPSc с аномальной третичной структурой.
Синдром впервые был описан в 1935 г. австрийскими неврологами Йозефом
Герстманом (Jozef Gerstmann), Эрнстом Штраусслером (Ernst Straussler) и Ильёй
Шейнкером (Ilya Scheinker) [1].
В 1986 г. был картирован ген PRNP, ассоциированный с прионными
заболеваниями, в том числе с развитием синдрома Герстмана – Штраусслера – Шейнкера
[2].
Распространенность и тип наследования
Начало заболевания, как правило, приходится на третье-четвертое десятилетия
жизни. Болезнь начинается с развития мозжечковой атаксии (нарушения согласованности
движений различных мышц при отсутствии мышечной слабости), затруднений при ходьбе
и удерживании равновесия, дизартрии (нарушения речи и произношения). Постепенно к
этим симптомам присоединяются прогрессирующие изменения личности и деменция.
Могут также отмечаться двоение в глазах, парез взора, глухота, пирамидные симптомы
(спастичность, непроизвольное резкое сокращение мышц конечностей в состоянии
пассивного движения). Значительно реже встречаются миоклонии (кратковременное
быстрое сокращение мышц), более характерные для болезни Крейтцфельдта – Якоба [6].
Первичную диагностику болезни проводит невролог. Далее назначается
электроэнцефалография, компьютерная томография и магнитно-резонансная томография
головного мозга. Для подтверждения диагноза возможно проведение молекулярно-
генетического анализа гена PRNP.
В настоящее время лечения, способного изменить ход болезни, не существует.
Поэтому, как правило, проводится симптоматическая терапия, с использованием
ноотропных лекарственных препаратов, протекторы когнитивных функций, корректоров
поведения и пр.
Средняя продолжительность жизни пациентов после начала болезни составляет 5
лет [4], в отличии от болезни Крейтцфельдта – Якоба, при которой большинство
пациентов умирают в первый год болезни, редко — в течение 2 лет и позже.
Синдром ГШШ, как и болезнь Крейтцфельдта – Якоба, ассоциирован с мутациями
гена PRNP, кодирующего нормальный белок PrPC, участвующий в формировании
миелиновой оболочки нервных волокон, и изоформу этого белка — прионный белок
PrPSc с аномальной третичной структурой [5].
Мутации вызывают конформационные изменения PrPC. Структура белка из
обогащенной α-спиральными участками превращается в PrPSc с β-складчатой структурой
[4]. Также способствует превращению обычного PrPС в патогенный инфекционная форма
белка PrPSc, попадающая в организм извне. Патогенная форма PrPSc, способная
превращать PrPС в PrPSc посредством белок-белковых взаимодействий, устойчива к
протеолизу и поэтому накапливается в тканях мозга больных в виде бляшек, содержащих
нитевидные или палочкообразные агрегаты этого белка. Результатом становится атрофия
таламических нейронов, что приводит к мозжечковым нарушениям (нарушения
координации движения, способности к пространственной ориентации), параличам,
экстрапирамидным нарушениям (двигательные нарушения, возникающие в результате
повреждения базальных ганглиев), глухоте, слепоте и деменции [5].
C заболеванием связан ген PRNP, мутации:
- c.305C>T (p.Pro102Leu)
- c.314C>T (p.Pro105Leu)
- c.350C>T (p.Ala117Val)
- c.593T>C (p.Phe198Ser)
- c.650A>G (p.Gln217Arg)
- Gerstmann J., Sträussler E., Scheinker I. Über eine eigenartige hereditär- familiäre
Erkrankung des Zentralnervensystems // Zeitschrift für die gesamte Neurologie und Psychiatrie.
– 1935. – V.154. – I. 1. – P. 736–762 - Sparkes R. S., Simon M., Cohn V. H., Fournier R. E. K., Lem J., Klisak I., Heinzmann C.,
Blatt C., Lucero M., Mohandas T., DeArmond S. J., Westaway D., Prusiner S. B., Weiner L. P.
Assignment of the human and mouse prion protein genes to homologous chromosomes. Proc.
Nat. Acad. Sci. 83: 7358-7362, 1986.[PubMed: 3094007] - Farlow M. R., Yee R. D., Dlouhy S. R., Conneally P. M., Azzarelli B., Ghetti B. Gerstmann-
Straussler-Scheinker disease. I. Extending the clinical spectrum. Neurology 39: 1446-1452,
1989.[PubMed: 2812321] - Завалишин И. А., Шиткова И. Е., Жученко Т. Д. Прионы и прионные болезни //
Клиническая микробиология и антимикробная химиотерапия. – 2000. – Т. 2. – №2. – С. 12-
19 - Prusiner S. B. Novel proteinaceous infectious particles cause scrapie. (1982) Science, 216,
136–144 - Н. Н. Заваденко, Г. Ш. Хондкарян, Р. Ц. Бембеева, А. А. Холин, Е. Н. Саверская.
Прионные заболевания человека: современные аспекты // Журнал неврологии и
психиатрии им. С.С. Корсакова. 2018;118(6): 88-95
Источник