
Синдром гурлера синдром шейе и синдром гурлера шейе

Генетика мукополисахаридозов. Наследование синдромов Гурлер, Шейе, Хантера
Мукополисахариды или гликозаминогликаны — полисахаридные цепи, синтезируемые клетками соединительной ткани как нормальный компонент многих тканей. Они состоят из большого числа дисахаридных блоков; отличительная черта конкретного гликозаминогликана — природа двух молекул сахаров.
Разложение этих макромолекул происходит в лизосоме и требует пошагового удаления блока моносахаридов в конце цепи с помощью фермента, специфичного для моносахарида и вовлеченной цепи. Таким образом, для разложения любого гликозаминогликана необходима серия ферментов, и один фермент часто участвует в распаде более чем одного гликозаминогликана.
Мукополисахаридозы — разнородная группа, включающая более дюжины болезней накопления, при которых в результате недостатка одного из ферментов, необходимого для их разложения, в лизосомах накапливаются мукополисахариды. При конкретных мукополисахаридозах могут накапливаться один или несколько гликозаминогликанов, если отсутствует фермент, необходимый для их распада.
Недеградировавшие гликозаминогликаны появляются в моче, где они могут быть обнаружены скрининговыми тестами.
Первые два описанных мукополисахаридоза — Х-сцепленный рецессивный синдром Хантера и более тяжелый аутосомно-рецессивный синдром Гурлер. Каждое из этих заболеваний сначала называли гаргоилизмом из-за грубости черт лица больных детей. Больные дети имеют скелетные аномалии и низкий рост, умственную задержку, а также другие аномалии.
Синдром Гурлер — следствие выраженной недостаточности а1-идуронидазы. Клинически отличное заболевание — синдром Шейе — первоначально считали вызванным мутацией в другом локусе, в основном из-за значительно более мягкого фенотипа. Тем не менее синдромы Шейе и Гурлер оказались аллельными, а мутации а1-идуронидазы, вызывающие синдром Шейе, связаны с более высокой остаточной активностью.
Примеры мукополисахаридозов:
1. Гурлер. Диагностируется рано (<18 мес), помутнение роговицы, скелетные изменения, гепатоспленомегалия, грубое лицо, смерть до 10-летнего возраста. Дефект фермента: a-1-идуронидаза. Наследование: аутосомно-рецессивное. Из-за аллелей, которые существенно нарушают активность фермента.
2. Шейе. Начало после 5 лет жизн, нормальный интеллект и продолжительность жизни, роговичные помутнения, клапанные пороки сердца. Дефект фермента: a-1-идуронидаза. Наследование: аутосомно-рецессивный. Очевидно, из-за аллелей, которые оставляют некоторую остаточную активность фермента.
3. Хантера. Гурлер-подобный синдром, но с медленным течением. Дефект фермента: Идуронатсульфатаза. Наследование: Х-сцепленный рецессивный. Мягкий фенотип с переменным поражением цнс.
Разные типы наследования аутосомного синдрома Гурлер и Х-сцепленного синдрома Хантера указывают, что они возникли вследствие мутаций в разных генах. Это различие также показано на клеточных культурах. Хотя фибробласты пациентов при обеих болезнях накапливают мукополисахариды в культуральной среде, накопление корректируется при совместном культивировании обоих типов клеток в одной культуральной посуде.
Коррекция происходит вследствие захвата фибробластами с недостаточностью a-L-идуронидазы при синдроме Гурлер нормальной a-L-идуронидазы, синтезированной фибробластами синдрома Хантера; в клетках с синдромом Хантера происходил обратный феномен. Этот простой эксперимент оказался красивой иллюстрацией того, что две болезни повреждают разные белки. Явление, когда продукт генома одного мутантного гена может скорректировать биохимический дефект у другого, называется генетической комплементацией, а исследования, используемые для обнаружения генетической комплементации, называют анализом комплементации.
Способность клетки усваивать недостающий лизосомный фермент из внеклеточной жидкости — один из механизмов коррекции биохимического дефекта при болезнях накопления после пересадки пациентам нормальных клеток (выделяющих фермент). Радикальный терапевтический эффект получен при лечении некоторых пациентов с мукополисахаридозами, включая синдром Гурлер, при пересадке костного мозга.
Способность клеток усваивать лизосомные ферменты из внеклеточной жидкости также дает обоснование для заместительной ферментной терапии при многих болезнях — стратегия, в значительном числе случаях оказавшаяся в высшей степени эффективной.
– Также рекомендуем “Генетика I-клеточной болезни. Молекулярные основы”
Оглавление темы “Генетика ферментопатий”:
- Генетика фенилкетонурии. Наследование
- Генетика болезни Тея-Сакса. Наследование
- Генетика мукополисахаридозов. Наследование синдромов Гурлер, Шейе, Хантера
- Генетика I-клеточной болезни. Молекулярные основы
- Генетика менделирующей восприимчивости к грибковой и бактериальной инфекции – mendelian susceptibility to mycobacterial disease (MSMD)
- Генетика гомоцистинурии. Наследование
- Генетика нарушений обмена витаминов (кофакторов). Наследование
- Генетика ферментопатий (ферментативной недостаточности). Наследование
- Генетика недостаточности альфа-1-антитрипсина. Наследование
- Генетика острой перемежающейся порфирии. Наследование
Источник
Мукополисахаридоз, тип I (Синонимы: недостаточность фермента лизосомной a-L-идуронидазы, синдромы Гурлер, Гурлер-Шейе и Шейе).
Мукополисахаридоз, тип I – аутосомно-рецессивное заболевание, возникающее в результате снижения активности лизосомной a-L-идуронидазы, которая участвует в метаболизме гликозаминогликанов. Заболевание характеризуется прогрессирующими нарушениями со стороны внутренних органов, костной системы, психоневрологическими и сердечно-лёгочными расстройствами.
Код по МКБ-10
- Е76 Нарушения обмена гликозаминогликанов.
- Е76.0 Мукополисахаридоз, тип I.
Эпидемиология
Мукополисахаридоз I – панэтническое заболевание с частотой встречаемости в популяции в среднем 1 на 90 000 живых новорождённых. Средняя частота синдрома Гурлер в Канаде 1 на 100 000 живых новорождённых, синдрома Гурлер-Шейе – 1 на 115 000, синдрома Шейе – 1 на 500 000.
Классификация
В зависимости от выраженности клинических симптомов заболевания различают три формы мукополисахаридоза I: синдромы Гурлер, Гурлер-Шейе и Шейе.
Причины мукополисахаридоза I типа
Мукополисахаридоз I – аутосомно-рецессивное заболевание, возникающее в результате мутаций в структурном гене лизосомной альфа-L-идуронидазы.
Ген альфа-L-идуронидазы – IDUA – расположен на коротком плече хромосомы 4 в локусе 4р16.3. К настоящему времени известно более 100 различных мутаций в гене IDUA. Превалирующее число известных мутаций – точечные в различных экзонах гена IDUA. Для европеоидов характерны две частые мутации Q70X и W402X.
Самая распространённая мутация среди пациентов из российской популяции – мутация Q70X. Её частота – 57%, что сравнимо с частотой Q70X в скандинавской популяции (62%). Частота мутации W402X, которая встречается в 48% случаев мукополисахаридоза I в ряде европейских популяций, в российской популяции составляет 5,3%.
Патогенез мукополисахаридоза I типа
Фермент a-L-идуронидаза участвует в метаболизме двух гликозаминогликанов – дерматан-сульфата и гепарансульфата. Поскольку идуроновая кислота входит в состав дерматансульфата и гепарансульфата, при данном заболевании нарушен внутрилизосомный распад именно этих гликозаминогликанов, которые и накапливаются в лизосомах повсеместно: в хрящах, сухожилиях, надкостнице, эндокарде и сосудистой стенке, печени, селезёнке и нервной ткани. Отёк мягкой мозговой оболочки вызывает частичную окклюзию субарахноидальных пространств, что приводит к прогрессирующей внутренней и наружной гидроцефалии.
Поражаются клетки коры большого мозга, таламуса, ствола, передних рогов. Тугоподвижность суставов – результат деформации метафизов, утолщение суставной капсулы вторично по отношению к отложению в ней гликозаминогликанов и фиброзу. Обструкция дыхательных путей – следствие сужения трахеи, утолщения голосовых связок, избыточности отёчных тканей в верхних дыхательных путях.
Симптомы мукополисахаридоза I типа
Мукополисахаридоз, тип IH (синдром Гурлер)
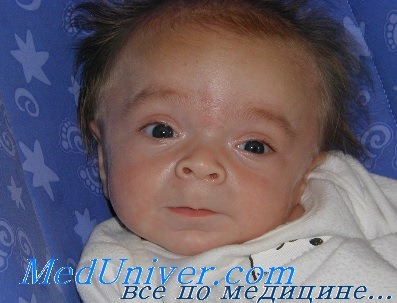
У больных с синдромом Гурлер первые клинические признаки заболевания появляются на первом году жизни, с пиком манифестации от 6 до 12 мес. В ряде случаев уже с рождения наблюдают незначительное увеличение печени, пупочные или пахово-мошоночные грыжи. Обычно диагноз устанавливают в возрасте от 6 до 24 мес. Характерные изменения черт лица по типу гаргоилизма становятся очевидными к концу первого года жизни: большая голова, выступающие лобные бугры, широкая переносица, короткие носовые ходы с вывернутыми наружу ноздрями, полуоткрытый рот, большой язык, толстые губы, гиперплазия дёсен, нерегулярные зубы. Другие частые манифестные симптомы – тугоподвижность мелких и крупных суставов, кифоз поясничного отдела позвоночника (поясничный гибус), хронические отиты и частые инфекционные заболевания верхних дыхательных путей. Практически у всех пациентов с синдромом Гурлер, так же как и при других типах мукополисахаридоза, кожные покровы плотные на ощупь. Часто встречается гипертрихоз. У единичных больных в возрасте до 1 года заболевание дебютировало с развития острой сердечной недостаточности, вызванной эндокардиальным фиброэластозом. По мере прогрессирования заболевания присоединяются симптомы, свидетельствующие о вовлечении в патологический процесс внутренних органов, сердечно-лёгочной, центральной и периферической нервной систем. Ведущие неврологические симптомы – снижение интеллекта, задержка речевого развития, изменения мышечного тонуса, сухожильных рефлексов, поражение черепных нервов, комбинированная кондуктивная и нейросенсорная тугоухость. Прогрессирующая вентрикуломегалия часто приводит к развитию сообщающейся гидроцефалии. К концу первого и в начале второго года жизни появляются шумы в сердце, позднее формируются приобретённые аортальные и митральные пороки сердца. К концу второго года жизни выявляют гепатоспленомегалию и характерные скелетные нарушения по типу множественного дизостоза: короткую шею, задержку роста, тотальную платиспондилию, поясничный гибус, тугоподвижность мелких и крупных суставов, дисплазию тазобедренных суставов, вальгусную деформацию суставов, изменение со стороны кистей по типу «когтистой лапы», деформацию грудной клетки в виде бочкообразной или колоколообразной. Часто наблюдается прогрессирующее помутнение роговицы, мегалокорнеа, глаукома, застойные диски зрительных нервов и/ или их частичная атрофия.
Ранние рентгенологические признаки – деформация рёбер (по типу «вёсельных») и овоидная деформация тел позвонков, излишняя трабекуляция диафизов длинных трубчатых костей в сочетании с её недостаточностью в области метафизов и эпифизов. По мере прогрессирования заболевания формируется макроцефалия с утолщением костей свода черепа, преждевременным закрытием лямбдовидного и сагиттального швов черепа, уменьшение орбит, расширение спинки турецкого седла. Больные погибают обычно в возрасте до 10 лет от обструкции дыхательных путей, респираторных инфекций, сердечной недостаточности.
Мукополисахаридоз, тип I-H/S (синдром Гурлер-Шейе) Клинический фенотип синдрома Гурлер-Шейе занимает промежуточное положение между синдромами Гурлер и Шейе, для него характерны медленно прогрессирующие нарушения со стороны внутренних органов, костной системы, лёгкое снижение интеллекта или отсутствие такового. Заболевание обычно дебютирует в возрасте 2-4 лет. Основные клинические нарушения – поражение сердца и развитие обструктивного синдрома верхних дыхательных путей. У некоторых пациентов наблюдают тотальный спондилолистез, что может приводить к компрессии спинного мозга. Большинство пациентов доживают до третьего десятилетия жизни. Основная причина летального исхода – острая сердечно-сосудистая и лёгочная недостаточность.
Мукополисахаридоз, тип IS (синдром Шейе)
В первоначальной классификации мукополисахаридозов, до открытия первичного биохимического дефекта при синдроме Шейе, его выделяли в отдельный тип – мукополисахаридоз V. Синдром Шейе – наиболее мягкий по течению заболевания среди других форм мукополисахаридоза I, для него характерны тугоподвижность суставов, аортальные пороки сердца, помутнение роговицы и признаки множественного костного дизостоза. Первые симптомы обычно появляются в возрасте от 5 до 15 лет. Ведущие клинические симптомы – скелетные нарушения в виде тугоподвижности суставов с развитием карпального туннельного синдрома. Офтальмологические расстройства включают помутнение роговицы, глаукому и пигментную дегенерацию сетчатки. Нейросенсорная тугоухость – позднее осложнение заболевания. Обструктивный синдром верхних дыхательных путей часто приводит к возникновению апноэ во время сна, что в некоторых случаях требует установления трахеостомы. Миелопатия шейного отдела спинного мозга встречается реже, чем при синдроме Гурлер-Шейе. Часто отмечают стеноз аорты с недостаточностью кровообращения и гепатоспленомегалию. Интеллект при данном синдроме не страдает или наблюдают лёгкие когнитивные нарушения.
Диагностика мукополисахаридоза I типа
Лабораторные исследования
Подтверждающая биохимическая диагностика мукополисахаридоза I заключается в определении уровня экскреции гликозаминогликанов мочи и измерении активности лизосомной a-L-идуронидазы. Суммарная экскреция Ггликозаминогликанов в моче возрастает. Также наблюдают гиперэкскрецию дерматансульфата и гепарансульфата. Активность a-L-идуронидазы измеряется в лейкоцитах или культуре кожных фибробластов с использованием искусственного флюорогенного или хромогенного субстратов.
Пренатальная диагностика возможна путём измерения активности a-L-идуронидазы в биоптате ворсин хориона на 9-11-й неделе беременности и/или определения спектра ГАГ в амниотической жидкости на 20-22-й неделе беременности. Для семей с известным генотипом возможно проведение ДНК-диагностики.
Функциональные исследования
При рентгенографии у пациентов с синдромом Гурлер обнаруживают типичные признаки так называемого множественного костного дизостоза. При МРТ головного мозга обнаруживают множественные кисты в перивентрикулярных областях белого вещества головного мозга, мозолистого тела, реже базальных ганглиев, признаки гидроцефалии; в редких случаях – пороки головного мозга в виде лиссэнцефалии, мальформации Денди-Уокера.
Дифференциальная диагностика
Дифференциальная диагностика проводится как внутри группы мукополисахаридозов, так и с другими лизосомными болезнями накопления: муколипи-дозами, галактосиалидозом, сиалидозом, маннозидозом, фукозидозом, GM1-ганглиозидозом.
Лечение мукополисахаридоза I типа
При синдроме Гурлер показана трансплантация костного мозга, которая может кардинально изменить течение заболевания и улучшить его прогноз, однако эта процедура имеет много осложнений и проводится на ранних стадиях заболевания, преимущественно в возрасте до 1,5 лет. В настоящее время создан препарат для ферментозаместительной терапии мукополисахаридоза I – альдуразим (Aldurazyme, Genzyme), который зарегистрирован в странах Европы, США, Японии; его применяют для лечения экстраневральных нарушений при мукополисахаридозе I. Препарат показан для коррекции мягких форм мукополисахаридоза I (синдромах Гурлер-Шейе и Шейе). Препарат вводят еженедельно, внутривенно, капельно, медленно, в дозе 100 ЕД/кг. Для лечения синдрома Гурлер с выраженными неврологическими осложнениями препарат менее эффективен, поскольку фермент не проникает через гематоэнцефалический барьер.
[1], [2]
Источник
Синдромы Гурлер, Гурлер-Шейе и Шейе – это одна болезнь, известная как мукополисахаридоз I (МПС I), кроме того, их называют МПС I H, МПС I H/S и МПС I S соответственно. Синдром Гурлер – одна из самых тяжелых форм и названа по имени Гертруды Гурлер, которая описала мальчика и девочку с данным заболеванием в 1919 г. В 1962 г. доктор Шейе, офтальмолог, наблюдал пациента с помутнением роговицы и очень мягкой формой синдрома. Заболевание, которое он описал, назвали синдром Шейе. Считалось, что синдром Шейе – это другая форма мукополисахаридоза (МПС) и отличается от синдрома Гурлер. Биохимические изменения при МПС I были выяснены в 1971 г., и обнаружили, что синдром Шейе и синдром Гурлер имеют одну и ту же причину – снижение активности фермента альфа-L- идуронидазы. Позже были описаны несколько случаев с промежуточными формами заболевания. Они не подходили ни к тяжелой, ни к легкой форме и были классифицированы как синдром Гурлер-Шейе. Сейчас стало известно, что МПС I очень разнообразны по своим клиническим проявлениям, несмотря на то, что имеют мутации в одном и том же гене.
При МПС I обычно наблюдается значительная задержка роста. Младенцы с синдромом Гурлер часто рождаются довольно крупными и в течение первого года растут быстрее нормы. Их рост замедляется к концу 1-го года и обычно останавливается в возрасте 3 лет. Они вырастают до 120 см. Рост при синдроме Гурлер-Шейе, как правило, не выше 152 см. Пациенты с синдромом Шейе обычно имеют нормальный рост.
Интеллект
При синдроме Гурлер происходит накопление ГАГ в клетках нервной системы, что является причиной замедления их развития в возрасте 1 — 3 лет и прогрессирующей потери приобретенных навыков в дальнейшем. В зависимости от тяжести заболевания степень неврологических и интеллектуальных нарушений различна: некоторые дети могут говорить лишь несколько слов, в то время как другие умеют хорошо говорить и немного читать. Они любят детские стихи, песенки и простые загадки. Очень важно помочь детям освоить как можно больше навыков и знаний до начала прогрессирования болезни. Даже когда дети начинают терять приобретенные навыки, они все еще продолжают понимать и радоваться жизни.
Пациенты с синдромом Гурлер имеют многочисленные проблемы со здоровьем, которые сильно препятствуют их обучению, – хронические отиты, снижение зрения, слуха, тугоподвижность суставов, гидроцефалию и нарушения дыхания. Соответствующее лечение этих проблем может существенно помочь в обучении таких детей.
При синдроме Гурлер-Шейе уровень интеллектуального развития нормальный, но у некоторых наблюдаются небольшие трудности с обучением. Кроме того, другие проблемы со здоровьем могут препятствовать обучению и общению. Пациенты с синдромом Шейе имеют нормальный уровень интеллектуального развития, но имеются данные о наличии психологических нарушений. Один из пациентов доктора Шейе имел очень высокий уровень интеллекта.
Внешний вид
На первом году жизни у ребенка резко изменяется форма черепа. Череп младенцев мягкий, и отдельные кости черепа разделены тонкими фиброзными тканями, которые называются швами. Впереди надо лбом и сзади около макушки находятся передний и задний роднички, мягкие места в черепе, которые закрываются в течение первых лет. При синдроме Гурлер шов вдоль верхней части головы соединяется раньше, чем положено, и поэтому череп вытягивается вперед и назад, становится вытянутым, и начинают выступать лобные бугры. Часто на лбу виден выступ, где череп закрылся преждевременно.
Из других внешних особенностей отмечается также широкий нос с плоской переносицей и широкими ноздрями. Глазные впадины неглубокие, поэтому глаза слегка выступают вперед. Язык увеличен и может вываливаться изо рта. Очень часто детям неправильно ставят диагноз гипотиреоза именно из-за измененного внешнего вида. Волосы у больных с МПС, как правило, очень жесткие и густые, иногда наблюдается повышенный рост волос на спине и руках.
Внешность людей с синдромом Шейе очень различна. Взрослые обычно коренасты, и их туловище короче конечностей, шея несколько укорочена. Пациенты с синдромом Шейе практически не имеют внешних особенностей.
Костная система
Со стороны костно-суставной системы при МПС I выявляется множественная симптоматика. У всех пациентов формируется тугоподвижность всех групп суставов, в результате контрактур межфаланговых суставов и укорочения фаланг, образуются деформации кистей по типу «когтистой лапы». Тазобедренные суставы сформированы неправильно, головки бедренных костей маленькие, уплощенные. У некоторых младенцев с МПС I наблюдается подвывих бедер. Эта проблема должна исправляться сразу же после рождения, так как позже с ней трудно справиться.
Походка многих пациентов с МПС I нарушена – они стоят и ходят с согнутыми коленями и бедрами. Иногда может наблюдаться Х-образное искривление ног, что обычно не требует лечения. Ступни ног широкие и негибкие, пальцы поджаты и скрючены. Подвздошные кости приобретают «треугольную» деформацию.
Рентгенологические изменения, видимые при синдроме Гурлер, описываются как множественный дизостоз. Ключицы укорочены, утолщены. Ребра описываются как «веслообразные». Фаланги кистей и стоп укорочены, имеют трапециевидную форму. Формируются кифоз, кифосколиоз. Кости позвоночника в нормальном состоянии выровнены по линии от шеи до ягодиц. При синдроме Гурлер и Гурлер-Шейе позвоночник может быть плохо сформирован, его позвонки не могут стабильно взаимодействовать друг с другом. Один или два позвонка в середине спины иногда бывают меньше, чем остальные, и слегка сдвинуты. Такое смещение позвонков может быть причиной развития искривления позвоночника (кифоз или горб). Однако обычно при заболевании искривление позвоночника довольно легкое и не нуждается в лечении.
Кости, которые стабилизируют соединения между головой и шеей, при МПС плохо сформированы, что делает шею нестабильной. В ряде случаев показано хирургическое вмешательство, чтобы соединить все косточки друг с другом. Если возникают сильные боли или слабость, или дрожь ног, нужно проводить исследование шеи (МРТ и рентген в согнутом и вытянутом положениях), чтобы оценить состояние шейных позвонков и спинного мозга.
Центральная нервная система
Прогрессирующие психические расстройства характерны для синдрома Гурлер, в то время как при мягких формах МПС I (синдромы Гурлер-Шейе и Шейе) интеллект больных практически не страдает или наблюдаются легкие когнитивные нарушения. Психомоторное развитие при синдроме Гурлер идет с заметным возрастным отставанием и достигает максимального развития на уровне 2-4 лет, затем останавливается и переходит (вместе с моторным развитием) в стадию регресса, достигая полной деменции. Однако систематические занятия, направленные на развитие когнитивных функций, способствуют более длительному сохранению интеллекта.
Поведенческие нарушения: регресс когнитивных функций наряду с тяжёлой потерей слуха, недостатком сна оказывают существенное влияние на поведение ребенка. По мере нарастания когнитивного дефицита к гиперактивности и агрессивности присоединяются аутистические черты.
Медикаментозная терапия, направленная на контроль разрушительного поведения, часто бывает неэффективной. Прогрессирующая сообщающаяся гидроцефалия является наиболее частым симптомом синдрома Гурлер и редко встречается при мягких формах МПС I типа (синдромах Гурлер-Шейе и Шейе).
При сдавлении спинного мозга, вызванного утолщением его оболочек, отмечают: нарушение походки, мышечную слабость, неуклюжесть при сохранных моторных навыках и дисфункцию мочевого пузыря. При тяжёлой форме заболевания часто наблюдаются судороги, что требует проведения оценки неврологического статуса. У пациентов со слабо выраженными клиническими признаками судорожный синдром встречается намного реже. Прогрессирование заболевания обычно хорошо поддается монотерапии антиконвульсантами.
Карпальный тоннельный синдром – частая нейропатия сдавления у пациентов в возрасте от 5 до 10 лет и взрослых. Люди с МПС I иногда испытывают боль и потерю чувствительности кончиков пальцев, вызванные запястным синдромом. Запястье состоит из восьми маленьких костей, которые соединены фиброзными связками, называемыми лигаментами. Нервы проходят через запястье между запястными костями и связками. Утолщение лигаментов оказывает давление на нервы. Такое повреждение нерва вызывает дистрофию мышц в основании большого пальца. При этом пациент испытывает трудности c захватом предметов.
Если ребенок жалуется на боль и онемение пальцев, особенно ночью, необходимо сделать исследование, называемое изучением нервной проводимости. Такое исследование может показать, является ли запястный синдром причиной таких симптомов. Если у ребенка наблюдаются какие-либо слабости руки или уменьшается мышечная масса у основания большого пальца, необходимо обследования у невропатолога.
Отсутствии лечения может привести к необратимой контрактуре дистальных межфаланговых суставов, а также к нарушению или потере чувствительности первых трех пальцев и парезу мышц тенара. К сожалению, пациенты редко сообщают о болевых ощущениях, пока не происходит потеря функции.
Головной и спинной мозг
Неврологические и интеллектуальные нарушения при синдроме Гурлер связаны с накоплением ГАГ в нейронах головного мозга. При синдромах Шейе и Гурлер-Шейе этого не происходит. Но следует не забывать, что другие нарушения могут вторично влиять на функции мозга – пониженный уровень кислорода, нарушения сна из-за апноэ, повышенное давление жидкости внутри и вокруг мозга (гидроцефалия головного мозга), нарушения слуха и зрения.
Головной и спинной мозг защищены от ударов с помощью спинномозговой жидкости, которая циркулирует вокруг них. У людей с МПС I циркуляция спинномозговой жидкости может быть заблокирована. Такая блокада (открытая гидроцефалия) вызывает повышение внутричерепного давления, что может привести к головным болям и задержке развития. Если возникают подозрения на гидроцефалию, то необходимо провести компьютерную томографию или МРТ. В случае подтверждения диагноза гидроцефалия лечится введением тонкой трубки (шунта), которая откачивает жидкость из головы в брюшную полость. У такого шунта есть чувствительный к давлению клапан, который позволяет откачивать цереброспинальную жидкость, когда давление вокруг мозга становится слишком высоким.
Органы зрения
Нарушения зрения, описанные здесь, являются общими для всех форм МПС I. Роговица может помутнеть из-за скопления ГАГ, что разрушает слои роговицы. Если помутнение роговицы тяжелое, то может ухудшиться зрение, особенно при тусклом свете. Некоторые пациенты не выносят яркого света, потому что помутнение вызывает неправильное преломление света. В этом случае может помочь ношение кепки с козырьком и солнечных очков. Многим пациентам с МПС I проводится трансплантация роговицы, что приводит к улучшению зрения.
Отложение мукополисахаридов в сетчатке может привести к потере периферического зрения и никталопии, или куриной слепоте. Ребенок может не хотеть ходить в темноте или просыпаться ночью и пугаться. Желательно оставлять ночник включенным в спальне и коридоре. Иногда могут возникать проблемы со зрением, вызванные изменениями в сетчатке глаза или глаукомой, то есть повышенным глазным давлением, что необходимо проверять во время регулярного осмотра у офтальмолога. Часто трудно определить, какие проблемы являются основной причиной ухудшения зрения. С помощью специальных исследований офтальмолог поможет определить, из-за чего происходит ухудшение зрения – из-за прохождения света (роговая оболочка) или из-за реакции глаза на свет (сетчатка или болезнь зрительного нерва).
Слух
Некоторая степень глухоты обычна для всех типов мукополисахаридозов. Это может быть кондуктивная глухота, глухота, связанная с поражением слухового нерва (нейросенсорная глухота), или комбинация обоих типов глухоты. Глухота усугубляется из-за частых ушных инфекций. Необходимо регулярно проводить проверку слуха (аудиометрия), для того чтобы сразу же начать лечение и максимально увеличить способность учиться и общаться. И нейросенсорная, и кондуктивная тугоухость в большинстве случаев могут компенсироваться подбором слуховых аппаратов.
Сердечно-сосудистая система
Характерно утолщение клапанов, сужение артерий, нарастающая ригидность миокарда, кардиомиопатии, артериальная гипертония. С возрастом может развиться сердечная недостаточность.
Практически у всех пациентов с МПС I наблюдается патология сердечно-сосудистой системы, которая характеризуется утолщением миокарда, снижением сократительной способности сердечной мышцы, изменениями со стороны клапанного аппарата и уплотнением створок и хорд клапанов, приводящими к формированию пороков сердца. Порок клапанов сердца может прогрессировать в течение многих лет без видимых клинических проявлений. Если состояние пациента ухудшается, требуется хирургическая операция для замены поврежденных клапанов. Поскольку проблемы с сердцем часто встречаются у пациентов c МПС I, с целью предупреждения возможных осложнений им необходимо делать эхокардиограмму каждый год.
При тяжелой форме синдрома из-за скоплений ГАГ может развиться поражение сердечной мышцы – кардиомиопатия. Сердце также может быть перегружено вследствие того, что приходится прокачивать кровь через измененные легкие, возникает аномальное увеличение правой стороны сердца или правосторонняя сердечная недостаточность. У некоторых пациентов наблюдается высокое кровяное давление.
Кожа
При МПС I кожа толстая и жесткая, что затрудняет забор крови и использование внутривенных катетеров. У некоторых наблюдается оволосение лица и спины. Обильное потоотделение, холодные руки и ноги являются обычным состоянием, что, возможно, связано с нарушениями со стороны сердечно-сосудистой системы или механизмами регулирования температуры тела. При появлении этих симптомов необходимо обследование кардиолога.
Проблемы с носом, горлом, грудью и ушами
У многих пациентов с МПС I отмечается шумное дыхание за счет отечности и утолщения слизистых верхних дыхательных путей и укорочения трахеи. Часто дети страдают хронической ринореей (насморком) вследствие анатомических особенностей строения носовых ходов (у них более короткие задние носовые ходы) и гипертрофии слизистых.
Миндалины и аденоиды часто увеличены и могут частично блокировать дыхательные пути. Шея обычно короткая, что способствует развитию проблем с дыханием. Трахея часто сужается из-за скопления слизи, часто более гибкая и мягкая из-за неправильного строения колец хряща в трахее. Форма груди у пациентов с МПС I неправильная, соединение между ребрами и грудиной не столь эластичное, поэтому грудь жесткая и не может двигаться свободно, в свою очередь, не позволяя легким набрать большой объем воздуха. Это становится причиной повторяющихся бронхитов и пневмоний.
Многие пациенты имеют шумное дыхание, даже когда нет никакой инфекции. Ночью у них может быть беспокойный сон и храп. Иногда у больного в течение коротких периодов могут наблюдаться остановки дыхания во сне (апноэ), такие остановки дыхания в 10 — 15 секунд не являются очень опасными, но это пугает родителей, поскольку они считают, что ребенок умирает. Если родители замечают у ребенка существенное нарушение ритма дыхания, удушье, они должны проконсультироваться со специалистом, который оценит состояние ребенка. В некоторых случаях апноэ лечится путем удаления миндалин и аденоидов, проведения вентиляции дыхательных путей при помощи непрерывного или двухуровневого положительного давления или проведения трахеостомии (операции по рассечению передней стенки трахеи с последующим введением в ее просвет канюли либо созданием постоянного отверстия – трахеостомы).
Многие семьи стараются избегать трахеостомии, так как эта мера является инвазивной и препятствует нормальной деятельности ребенка. На самом деле многие врачи считают, что пациенты с МПС I должны делать трахеостомию раньше, чем обычно это происходит. Такие пациенты чувствуют себя намного лучше после улучшения дыхания во время сна.
Желудочно-кишечная система
Гепатоспленомегалия при синдромах Гурлер и Гурлер-Шейе (печень и селезенка увеличены из-за накопления мукополисахаридов)). При синдроме Шейе печень также может быть увеличена. Увеличение печени обычно не приводит к нарушению ее функции, но может влиять на переносимость той или иной пищи, а также может способствовать ухудшению функции дыхания.
При МПС I живот увеличен в размерах из-за специфической осанки, слабости мышц и увеличенных печени и селезенки. Характерными являются дефекты передней брюшной стенки в виде сочетанных или изолированных грыж (пупочной, паховой, пахово-мошоночной и вентральной). Паховую грыжу необходимо оперировать, но иногда она возникает после операции вновь. Пупочные грыжи обычно не оперируют, вмешательство производят только в случаях, если происходит ущемление петли кишечника или грыжа становится очень больших размеров.
У больных с МПС I нередко наблюдается неустойчивый стул. При формах Гурлер-Шейе и Шейе часто возникают боли в животе. Диарея может исчезать с возрастом, но во время приема антибиотиков может появляться вновь. Хронические запоры появляются с возрастом, поскольку ребенок c МПС I становится менее активным и его мышцы ослабевают.
Источник