
Синдром клиппеля фейля характеризуется на рентгенограммах признаками

А.А. Устинович, Т.М. Галица, Е.Е. Жеребилова, С.В. Кузьмина, Г.В. Леус, А.Н. Ракуть
СИНДРОМ КЛИППЕЛЯ-ФЕЙЛЯ
Белорусский государственный медицинский университет,
УЗ 3-я городская детская клиническая больница
A.A. Ustinovich, T.M. Galitsa, E.E. Jerebilova, S.V. Kuzmina, G.V. Leus, A.N. Racut.
KLIPPEL – FEIL SYNDROME
В 1912 году французские врачи М. Klippel (невропатолог) и Andre Feil (рентгенолог) описали врожденный порок развития позвоночника, характеризующийся деформацией (укорочением) шеи, обусловленной уменьшением числа шейных позвонков, их сращением или меньшими размерами.
Различают три типа деформации: первый тип – уменьшение общего числа шейных позвонков; второй тип – синостоз всего спаянного в единую кость шейного отдела позвоночника с затылочной костью и верхними грудными позвонками; сочетание 1 или 2 типа с синостозом нижнегрудных и поясничных позвонков. Часто деформация сочетается с незаращением дужек позвонков (spina bifida cervicalis), наличием шейных ребер, синхондрозом лопаток с позвоночником при высоком их стоянии (болезнью Шпренгеля).
В большинстве случае синдром спорадичен, имеются данные о его генетической гетерогенности, например, 2 тип наследуется аутосомно-доминантно, а 3 тип – аутосомно-рецессивно.
У больных отмечается укорочение и ограничение подвижности шеи, низкая граница роста волос на затылке, кифосколиоз. Укорочение шеи придает пациентам особый вид – «человека-лягушки». В тяжелых случаях подбородок упирается в грудину, мочки ушей касаются плеч, затрудняется дыхание и глотание. У части больных могут быть крыловидные складки шеи, пороки развития мышц плечевого пояса. Лопатки широко разведены, часто укорочены. В большинстве случаев деформация безболезненна, но иногда сопровождается синдромом сдавления шейных корешков спинного мозга. Возможны асимметрия лица, аномалии зубов, микроцефалия, гидроцефалия, спинно-мозговая грыжа, пороки ребер, лучевой кости и ее производных, постаксиальная полидактилия. В 45% случаев наблюдаются гипоплазия и дистопия почек, в 25% – глухота, в 17 – 20% – расщелина неба, в 15% – пороки сердца. Также характерны пороки развития нервной системы и умственная отсталость. Со стороны глаз наблюдаются паралитическое косоглазие, гиперметропия, нистагм, синдром Горнера и Щтиллинга-Тюрка-Дуана. Также характерны: слабость рук и ног, переходящая позднее в спастические и паралитические параплегии и тетраплегии, нарушения функций симпатического отдела нервной системы, зеркальные движения конечностей (из-за возможного отсутствия перекреста пирамид), глухота, эпилептические припадки, приступы головной боли.
Диагностика синдрома основана на триаде клинических симптомов: укорочение шеи, наблюдаемое с рождения, низкая граница роста волос на шее и ограничение подвижности головы. Для уточнения типа деформации проводят рентгенологическое исследование шейного и грудного отделов позвоночника в прямой и боковой проекциях. На рентгенограммах чаще выявляют сращение 4-6 шейных позвонков в сплошную малодифференцированную костную массу. Иногда тела позвонков сливаются лишь частично и тогда можно проследить узкие полоски просветления – недоразвитые межпозвоночные диски. При полном синостозе блокированными оказываются тела, дужки и отростки позвонков. Частичный синостоз вызывает в процессе роста искривление позвоночника в сагиттальной или фронтальной плоскости.
Дифференциальный диагноз проводят с туберкулезным спондилитом верхних шейных позвонков, двусторонней и односторонней формами мышечной кривошеи (особенно при отсутствии эффекта от консервативного лечения).
Лечение, как правило, консервативное (ЛФК, массаж), направлено на улучшение осанки и предупреждение вторичных деформаций. При возникновении компрессии корешков спинного мозга, например шейными ребрами, их резецируют. Для устранения болевого синдрома назначают мягкий воротник типа Шанца, анальгезирующие средства, физиотерапию.
Витальный прогноз благоприятный при отсутствии пороков внутренних органов, однако у больных возникают серьезные косметические и функциональные проблемы.
В приводимом клиническом наблюдении выявлено редкое сочетание синдрома Клиппеля – Фейля и тератомы спинного мозга.
Девочка П. родилась от 1 беременности, 1 родов, в сроке 40 недель. Беременность отягощена хронической фето-плацентарной недостаточностью, в родах – раннее излитие околоплодных вод., угрожающий разрыв промежности, безводный период . 2 ч. 50мин. Масса тела при рождении – 3900, длина – 55 см, оценка по шкале Апгар 8/9 баллов. Выписана домой на 3 сутки жизни. На следующий день (4 сутки жизни) на врачебном патронаже отмечены жалобы на снижение аппетита, вялость, осиплость голоса, отсутствие движений в правой ручке и с диагнозом «ОРИ, перелом ключицы?» направлена в стационар.
С целью уточнения характера патологии ребенку выполнены обзорная рентгенограмма скелета, (рис.1), КТ и МРТ ЦНС.
Заключение КТ головного мозга: выявляется отек белого вещества головного мозга, умеренно выраженная внутренняя гидроцефалия Гипотрофия червя мозжечка. Кистоподобный тяж в проекции продолговатого мозга.
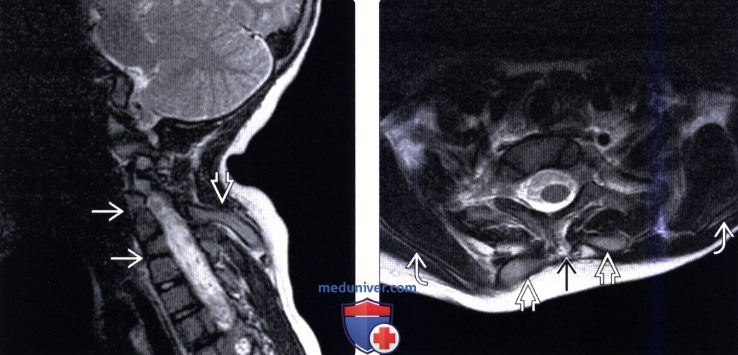
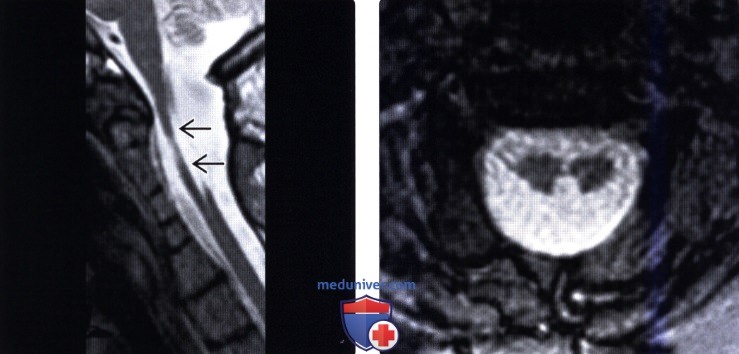
КТ шейного и грудного отдела позвоночника: врожденный костный блок тел 2.3.7. грудных позвонков с передней расщелиной 3 и 4 грудных позвонков. Поперечные отростки С5, С6, С7 позвонков слиты к костные конгломераты с обеих сторон. Аномалия сращения 3, 4 и 5 ребра с обеих сторон. Правая ключица вогнута в средних отделах. Широкая расщелина задней стенки позвоночного канала на протяжении С6 – Тh4, максимальной шириной до 21мм в коронарной плоскости. В проекции позвоночного канала на участке С5 -Тh2 определяется интрадуральное изогиперинтенсивное объемное образование 20×15мм, вызывающее деформацию (вогнутость) тел прилежащих позвонков: Заключение: КТ признаки объемного патологического образования позвоночного канала на участке С5 – Тh2 позвонков. Врожденные костные аномалии.
МРТ шейного отдела позвоночника: интрамедулярно на уровне С5 – Тh2 определяется объемное образование 20×15мм с бугристыми контурами, интенсивно накапливающее контрастное вещество, Имеет место врожденная аномалия спинного мозга. С уровня продолговатого мозга он раздвоен, истончен, диаметр 1,5×2 мм до уровня вышеописанного патологического образования. Гипоплазированы обе гемисферы мозжечка, его червь. В грудном отделе спинного мозга на уровне Тh7, Тh8, Тн12 -L1 определяется гипоинтенсивный сигнал (нельзя исключить сирингомиелию). Заключение: МРТ проявления интрамедуллярного объемного образования на уровне С5 -Тh2 спинного мозга. Врожденная аномалия спинного мозга (его раздвоенн6ость в шейном отделе) Сирингомиелия в грудном отделе, гипоплазия червя мозжечка, его гемисфер.
На 8 сутки жизни ребенок был переведен в нейрохирургическое отделение 9 городской клинической больницы с диагнозом: Объемное образование спинного мозга (зрелая тератома) на уровне С7 -Тн3 позвонков. ВПР ЦНС: болезнь Клиппеля – Фейля с гипоплазией мозжечка. Тетрапарез, больше справа. Бульбарный синдром.
На 11 сутки жизни ребенку была проведена ляминэктомия С7 – Тh3, тотальное удаление опухоли.
Гистологическое заключение: зрелая тератома.
В послеоперационный период в неврологическом статусе сохранялись бульбарные нарушения в виде нарушения глотания, тетрапарез, глубже справа, сохранялся длительный субфебрилитет.
Получала медикаментозное лечение: аналгин, цефатоксим, амикацин, аугментин, биофлор, дексаметазон, преднизолон, этамзилат, диалакт, панкреатин, инфузии СЗП, эритроцитной массы.
В дальнейшем, на 3-м месяце жизни у ребенка развился эпилептический синдром с наличием генерализованных припадков. На фоне респираторных проблем, обусловленных деформацией грудной клетки (рис. 2) и неврологической симптоматикой, сформировались кардиомегалия и легочная гипертензия II степени, НКI степени. В дальнейшем ребенок страдал глубокой задержкой психомоторного развития, также у него сохранялся судорожный синдром.
Таким образом, в данной клинической ситуации наблюдается редкое сочетание синдрома Клиппеля – Фейля с тератомой шейно-грудного отдела позвоночника, что обусловило развитие у ребенка тяжелых неврологических и соматических нарушений.

Источник
Синдром Клиппеля-Фейля (синоним: синдром короткой шеи) — редкий врожденный порок развития шейных и верхнегрудных позвонков, который характеризуется наличием у больного короткой и малоподвижной шеи. Данная патология наследственная, передается по аутосомно-доминантному типу. Впервые заболевание было описано французскими врачами Морисом Клиппелем и Андре Фейлем в 1912 году.
Выделяют две формы данной патологии:
- Порок, характеризующийся уменьшением общего числа шейных
- Синостоз костей шейного отдела позвоночника.
Особенности заболевания
Под синдромом Клиппеля-Фейля понимается врожденная патология шейного отдела, заключающаяся в синостозе и уменьшении числа позвонков. Она характеризуется высокой частотой проявления — один случай на 120 тысяч новорожденных. Наиболее типичным признаком заболевания считается выраженное уменьшение длины шеи. В большинстве случаев сопровождается иными аномалиями костно-мышечного аппарата и врожденными патологиями внутренних органов. В диагностике недуга принимают участие сразу несколько узких специалистов: невролог, ортопед, генетик и др. Консервативное лечение включает в себя ЛФК, физиотерапию и массаж. В особо серьезных случаях требуется хирургическое вмешательство — операция цервикализации.

Причины возникновения синдрома
Данный синдром входит в группу генетически детерминированных заболеваний. Изменения патологического характера начинают развиваться в первые недели развития плода в утробе. Врачи отмечают несколько основных причин, в результате которых синдром проявляется. К ним относится нарушение сегментации и развития позвоночного столба, в основном, на верхнешейковом уровне.
Классификация
По типу деформации позвоночника в шейном отделе болезнь Клиппеля-Фейля можно разделить на три вида: При первом типе деформация шейного отдела наступает по причине меньшего количества сформированных шейных позвонков (4-5 штук вместо положенных семи). При втором типе патология возникает вследствие уменьшения размеров позвонков шеи в сравнение с остальными частями позвоночника. При третьем типе наблюдается синостоз позвонков, то есть сращение шейных позвонков между собой, а также с затылочной костью и с позвонками грудного отдела в «монолитное» образование. Также возможно и сочетание двух или трех типов.
Симптомы
Клиническая картина синдрома Клиппеля-Фейля заметна сразу же после рождения больного ребенка:
- визуально шея выглядит короткой (голова словно лежит на плечах);
- волосяной покров растёт аномально низко;
- движения шеи ограничены, особенно, трудно даётся вращательная амплитуда.
Около трети случаев характеризуются другими специфическими признаками:
- искривление позвоночника;
- врождённая кривошея в костной форме (голова ребёнка нехарактерно повёрнута вбок);
- имеются кожные складки на шее; болезнь Шпренгеля (аномально высокое расположение лопаток у пострадавшего);
- различные дефекты строения/развития верхних конечностей;
- иногда встречаются аномалии развития нижних конечностей (искривление голеней, плоскостопие).
Обратите внимание! Внешние проявления заболевания не оказывают никакого влияния на психическое и физическое развитие ребёнка (в большинстве случаев). Очень опасными считаются патологии внутренних органов и систем, которые часто сопровождают синдром короткой шеи.
Часто встречаются следующие аномалии:
- страдает сердечно-сосудистая система (декстрапозиция аорты, патология межжелудочковой перегородки, незаращение боталлова протока);
Со стороны мочевыделительной системы отмечаются следующие аномалии:
- отсутствие или недоразвитие почки, несвойственное расположение устья мочеточника, накопление в почке жидкости, что опасно для жизни).
Отдельные случаи характеризуются отсутствием какого-либо органа (часто почки или мочеточника). Патология может угрожать жизни новорождённого. В большинстве ситуаций такие аномалии выявляют во время течения беременности на плановом УЗИ.

Диагностика
Диагноз ставится на основании данных осмотра, дополненных результатами исследований.
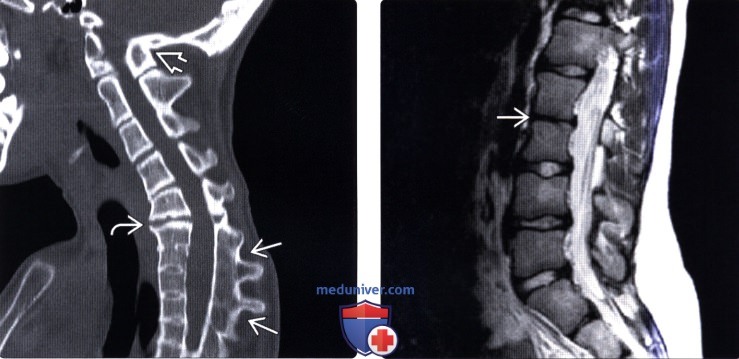
Для определения характера изменения позвонков основным методом является рентгенография. Ее проводят обязательно в 2 проекциях, из которых нередко более информативной является боковая. Это связано с тем, что из-за патологического положения головы тени от черепа накладываются на изображение позвоночника, затрудняя выявление деталей. Рекомендуется дополнительно сделать снимки в положениях максимального сгибания и разгибания шеи. Это поможет выявить возможную нестабильность тех позвонков, которые не срослись.
При рентгене шейного и грудного отделов позвоночника при синдроме Клиппеля-Фейля могут быть выявлены:
- деформированные, уменьшенные позвонки;
- уменьшение числа позвонков;
- срастание вместе тел позвонков, при этом может сформироваться как единый костный конгломерат, так и костная структура с хрящевыми включениями в виде недоразвитых межпозвоночных дисков;
- искривления позвоночного столба с образованием сколиоза, кифоза или лордоза;
- наличие дополнительных шейных ребер, синхондроз и аномальное стояние лопаток.
Дополнительно проводят диагностический поиск для выявления различных аномалий строения внутренних органов. Проводят ЭКГ, УЗИ почек и сердца.
При наличии неврологических осложнений может потребоваться УЗДГ сосудов шеи, ангиография, миелография, ЭМГ, ЭЭГ, КТ и МРТ шейного отдела позвоночника.
Обязательна консультация медицинского генетика, сбор семейного анамнеза, возможно проведение генетического исследования. Это необходимо для установления типа наследования и определения рисков для будущих поколений, что особенно актуально при планировании беременности.
Лечение
синдрома Клиппеля-Фейля заключается в таких мероприятиях:
- курс массажа;
- лечебная физкультура;
- применение воротника Шанца;
- физиотерапевтические процедуры;
- медикаментозное лечение;
- оперативное лечение.
Медикаментозное лечение направлено на устранение симптоматики. Из лекарственных препаратов предпочтение отдается анальгетикам, так как нестероидные противовоспалительные препараты не могут устранить болевой синдром.
В некоторых случаях лечебная физкультура, массаж, физиотерапевтические процедуры дают положительные результаты, однако при сильных корешковых болях медики прибегают к оперативному вмешательству. Выполняется операция, которая имеет название «цервикализация» по Бонола.
Проводится в несколько этапов:
- Проводится паравертебральный разрез между краем лопатки и остистыми отростками. Отсекаются мышцы от края лопатки, которые отводятся вовнутрь, а лопатка кнаружи.
- Затем удаляются первые четыре ребра и надкостница.
Оперативное вмешательство проводится на одной стороне, после окончания периода восстановления — на другой. Однако полностью излечить заболевание у людей с синдромом Клиппеля-Фейля невозможно, так как лечение направлено на предупреждение прогрессирования вторичных деформаций. Аномалия имеет относительно благоприятный прогноз, так как, по мнению специалистов, отклонения не слишком деструктивно сказываются на функциональной способности систем организма.
Лечение и спорт помогают поддерживать состояние здоровья на положительном уровне. Синдром Клиппеля-Фейля может развиваться и в негативную сторону, в таком случае подвижность головы будет постепенно ограничиваться, что может способствовать прогрессированию сложных неврологических аномалий.
Осложнения и прогноз для больного
После хирургического вмешательства и реабилитационного периода больной может вести полноценную жизнь. Шея за это время немного увеличится в длине, что, с эстетической точки зрения, считается благоприятным аспектом. Если пациент игнорирует рекомендации врача по восстановлению, организм начинает «отвечать» осложнениями. В первую очередь развиваются патологии внутренних органов, появляются сильные боли в области шеи. Последние обусловлены постоянным ущемлением нервных корешков и могут привести к полному обездвиживанию конечностей. Поражение внутренних органов опасно необратимыми патологическими процессами и может выступать причиной ранней смерти.
Загрузка…
Источник
Лучевая диагностика аномалии Клиппеля-Фейляа) Терминология: б) Визуализация: 1. Общие характеристики аномалии Клиппеля-Фейля: 2. Рентгенологические данные: 3. КТ признаки аномалии Клиппеля-Фейля: 4. МРТ признаки аномалии Клиппеля-Фейля: 5. Рекомендации по проведения исследования:
в) Дифференциальная диагностика аномалии Клиппеля-Фейля: 1. Ювенильный идиопатический артрит: 2. Послеоперационный костный блок: 3. Отдаленные последствия дисцита: 4. Анкилозирующий спондилит: г) Патология: 1. Общие характеристики: 2. Стадирование, степени и классификация аномалии Клиппеля-Фейля: 3. Макроскопические и хирургические характеристики: 4. Микроскопия: д) Клинические особенности: 1. Клиническая картина аномалии Клиппеля-Фейля: 2. Демография: 3. Течение заболевания и прогноз: 4. Лечение: е) Диагностическая памятка: ж) Список использованной литературы: – Также рекомендуем “Рентгенограмма, МРТ при нарушении формирования позвонков” Редактор: Искандер Милевски. Дата публикации: 18.7.2019 |
Источник