
Синдром леопарда что это такое

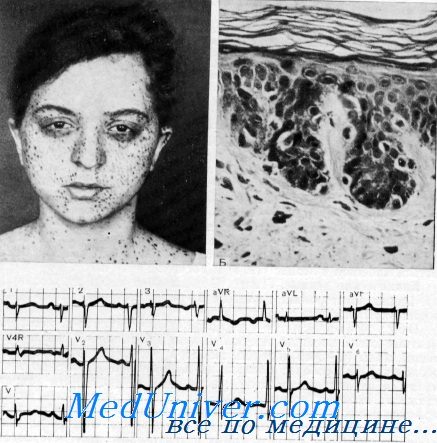
Синдром LEOPARD – клиника, диагностика, лечениеСинонимы: лентигиноз врожденный системный, лентигиноз кардиомиопатический, синдром Мойнахана (вариант синдрома LEOPARD, включающий дополнительно митральный стеноз), синдром Кейпюта—Римойна—Конигсмарка (вариант синдрома LEOPARD, описанный как сочетание лентигиноза с пороками сердца, врожденной глухотой, олигофренией, дизрафическим статусом в виде частичной синдактилии, расщепления задних отростков нижних грудных и верхних поясничных позвонков). Определение. Наследственное заболевание, характеризующееся нарушением пигментации в сочетании с поражением ЦНС, внутренних органов и опорно-двигательного аппарата. Историческая справка. В 1936 г. Е. P. Zeisler и S.W. Becker впервые описали синдром с наличием нескольких лентиго, гипертелоризма, деформации грудной клетки и прогнатизма. В 1966 г. R.J. Walther, B.J. Polansky и LA. Gratis описали семью, в которой мать, сын и дочь с генерализованным лентиго имели электрокардиографические аномалии. Наиболее полное описание синдрома выполнили в 1969 г. R.J. Gorlin и соавторы. Они же предложили назвать данный синдром акронимом LEOPARD из начальных букв основных симптомов заболевания (L: Lentigines — лентиго; Е: Electrocardiographic conduction abnormalities — электрокардиографические нарушения проводимости; О: Ocular hypertelorism — глазной гипертелоризм; Р: Pulmonary stenosis — стеноз легочной артерии; А: Abnormalities of genitalia — аномалии половых органов; R: Retardation of growth — задержка роста; D: Deafness sensorineural — нейросенсорная глухота). Этиология и патогенез синдрома LEOPARD. Заболевание генетически неоднородное. В 90% случаев обнаружены мутации гена PTPN11 (локус 12q24.1), в остальных случаях — мутации генов RAF1 (локус 3р25) и BRAF. Первичный продукт гена PTPN11, фермент тирозинфосфатаза, участвует в процессах внутриклеточной передачи сигналов и регуляции ответа клеток на внеклеточные стимулы. Недостаточность функции фермента приводит к изменению пролиферации клеток, нарушению их дифференцировки и жизнеспособности. Первичный продукт гена RAF1 относят к семейству протеинкиназ. Он участвует в защите клеток от гибели путем апоптоза. Активированный ген RAF1 обладает свойствами онкогена. Наследование аутосомно-доминантное, но синдром может возникнуть в результате спонтанной мутации. Частота. В настоящее время нет точных данных о количестве пациентов с этим синдромом, до 2011 г. в медицинской литературе описано чуть более 100 случаев. Возраст и пол. Чаще встречается у людей мужского пола. Симптомы, характерные для этого заболевания, можно обнаружить у ребенка в первые месяцы жизни. Однако существует мнение, что лентиго обнаруживается при рождении, а внекожные проявления — после полового созревания. Поражения кожи. Вначале обнаруживаются пятна цвета «кофе с молоком». Они обычно существуют с рождения или появляются в первые месяцы жизни. Данные изменения кожи диагностируются в 75% случаев у пациентов с этим синдромом. Лентигинозные элементы возникают, как правило, с 4—5-летнего возраста, их количество может быть значительным. В некоторых случаях у пациентов с данным синдромом лентиго отсутствует. Лентигинозные элементы имеют темно-коричневую окраску (иногда даже черную). Пигментные пятна темнее обычных веснушек, круглой или овальной формы, размерами 1—5 мм в поперечнике. Лентигинозные очаги чаще всего располагаются на коже шеи, туловища, реже — на лице, волосистой части головы, ладонях, подошвах и гениталиях. Слизистые оболочки не поражаются. С возрастом их количество нарастает и пигментация усиливается. Под влиянием УФО лентиго изменениям не подвергается. Иногда одновременно наблюдаются койло- или лейконихия.
Изменения костной системы. Лицо больных треугольной формы с широко расставленными глазами (гипертелоризм), с явлениями прогнатии и низко расположенными ушными раковинами. Возможны асимметрия черепа, сглаженность затылочного бугра, выступающие теменные бугры, микроцефалия. На рентгенограммах черепа выявляются деминерализация костей, пальцевидные вдавления, аномалии турецкого седла, неполное заращение швов, асимметрия придаточных пазух. Отмечают задержку роста и разнообразные аномалии скелета: килевидная или воронкообразная грудная клетка, крыловидные лопатки, кифоз, сколиоз, spina bifida, сращение костей шейного отдела позвоночника, гипертрофия лонных костей, гипермобильность суставов, подвывих и деформация головки бедренной кости, остеопороз различных отделов, «конская стопа». Изменения мочеполовых органов при синдрома LEOPARD. У одной трети пациентов мужского пола с данным синдромом отмечается односторонний или двухсторонний крипторхизм. Другие аномалии (гипоспадия, дисплазия яичек, позднее появление менструаций, дефекты яичников) наблюдаются редко. Изменения сердечно-сосудистой системы. Изменения ЭКГ могут быть обусловлены стенозом легочной артерии или субаортальным стенозом, дефектом межжелудочковой перегородки и, как результат этой патологии, гипертрофической кардиопатией. Выявляются нарушения внутрижелудочковой проводимости в виде удлиненного интервала PQ, расширения комплекса QRS. Возможны изменения зубца Р (двухфазный, отрицательный, остроконечный). Ось сердца обычно ориентирована между 60° и 120°. Другие изменения. Нейросенсорная глухота с вторичными дефектами речи и в 30% случаев небольшое отставание в умственном развитии. Выявляются отклонения в ЭЭГ, замедление проводимости в периферических нервах. Диагноз синдрома LEOPARD. В 1976 г. D. A. Voron и соавторы предложили два варианта критериев для диагностики данного синдрома: несколько лентиго плюс два других основных симптома; при отсутствии лентиго три основных симптома, а также наличие родственника с данным синдромом. Диагноз может быть установлен уже в первые месяцы жизни (по данным некоторых авторов, через 7,5±3,96 месяца от рождения). Диагноз устанавливался в этих случаях по трем основным клиническим признакам: характерные черты лица (гипертелоризм, неполное заращение швов, птоз, присутствие в 100%), кардиомиопатия (в 87%), пятна цвета «кофе с молоком» (в 75%). Синдром подтверждается обязательно молекулярно-генетическими исследованиями. Дифференцируют синдром LEOPARD с другими лентигинозами. Течение и прогноз. Существуют полные и неполные клинические формы синдрома. Наиболее серьезными являются изменения скелета и сердца. Лентигиноз доставляет только косметические проблемы. Лечение синдрома LEOPARD. Пациенты нуждаются в лечении стеноза легочной артерии, обструктивной кардиоми-опатии, дефектов межпредсердной перегородки, первичной легочной гипертензии, деформации грудной клетки, кифосколиоза, крипторхизма, умственной отсталости и тугоухости. – Также рекомендуем “Синдром Карни (NAME, LAMB) – клиника, диагностика, лечение” Оглавление темы “Меланозы”:
|
Источник
Синдром LEOPARD является комплексом дисморфогенетических расстройств с переменной пенетрантностью и экспрессивностью. Горлин впервые представил аббревиатуру LEOPARD в качестве названия этого синдрома в 1969 году. Буквы этой аббревиатуры описывают основные черты этого расстройства, а именно:
- Лентиго (L)
- Электрокардиографические нарушения проводимости (E)
- Глазной гипертелоризм (O)
- Стеноз легочной артерии (P)
- Аномалии половых органов (A)
- Замедление роста (R)
- Глухота (D)
Зейслер и Беккер первыми описали этот синдром в 1936 году, провев обследование 24-летней женщины с прогрессивным лентиго, гипертелоризмом и прогнатизмом. Вскоре были зафиксированны первые семейные случаи у близнецов Розен, а затем еще 8 человек из большой семьи. Монахан был первым, кто составил документальную ассоциацию синдрома с сердечными аномалиями и низкорослостью в 1962 году.
Синдром LEOPARD. Причины
Молекулярные исследования показали, что синдром LEOPARD является аллельным расстройством, которое вызывается различными мутациями в гене PTPN11, кодирующем протеин-тирозин-фосфатазу SHP-2, этот ген расположен в полосе 12q24.1. В 2005 году, Огата и Йошида документально подтвердили, что мутации в гене PTPN11 могут быть идентифицированы приблизительно у 80% больных с синдромом LEOPARD.
Еще один интересный случай синдрома LEOPARD был обнаружен в одной семье, у отца и его взрослого сына. Отец имел лентиго, которое было одинаково распределено по всему телу, в то время как у его сына лентиго отсутствовало на левой части грудной клетки, на спине, на левой и правой руках. Позже было установленно, что сын имел мозаичный кариотип в лимфоцитах (47, XXY / 46XY). При анализе тканей полученных на биопсии пигментированной кожи, в основном прослеживался кариотип 47, XXY в то время как кариотип 46, XY прослеживался в непигментированных областях. Некоторые исследователи пришли к выводу, что такие факторы как мозаичность, типы мутаций в гене PTPN11, а также кол-во и характеристики половых хромосом могут влиять на фенотип синдрома LEOPARD.
Синдром LEOPARD. Фото
Лентиго на лице ребенка с синдромом LEOPARD.
Лентиго на склере у ребенка с синдромом LEOPARD.
Неупорядоченная пигментация на туловище у пациента с синдромом LEOPARD.
Ониходистрофия
Синдром LEOPARD. Симптомы и проявления
Высоко вариабельная экспрессивность синдрома делает его диагностику трудной задачей, особенно в спорадических случаях. Семьдесят процентов случаев являются семейными. На основании клинического анализа большой серии пациентов, собранных в медицинской литературе, в 1976 Ворон вывел минимальные критерии для диагностики. Для синдрома LEOPARD обязательно наличие лентиго и по крайней мере двух из следующих проявлений и знаков:
- Другие кожные нарушения
- Сердечные структурные аномалии
- Аномалии в мочеполовой системе
- Эндокринные нарушения
- Неврологические дефекты
- Дисморфизмы лица
- Дефицит роста
- Скелетные аномалии
Если лентиго отсутствует, то диагноз синдрома LEOPARD можно поставить при наличии минимум трех из вышеуказанных знаков и проявлений. Диагностика синдрома LEOPARD сильно затрудненна у маленьких детей. Диагноз может быть клинически подозреваемым в первые месяцы жизни у пациентов, имеющих минимум три основных проявления: характерные черты лица (100%), гипертрофическая кардиомиопатия (87%) и кожные пятна цвета кофе с молоком (лентиго [75%]) .
Физическое обследование
Лентиго – мелкие, темно-коричневые, полигональные, неправильной формы пятна, размером, как правило, 2-5 мм в диаметре, но иногда и больше, до 1-1,5 см. Эти пятна часто присутствуют на лице, шее и на верхней части части туловища, на ладонях, ступнях и на склерах. Лентиго являются наиболее заметным проявлением синдрома LEOPARD. Оно присутствует у более чем 90% пациентов. Однако, отсутствие этих пятен не исключает наличия этого синдрома. При тщательном обследовании кожи, можно обнаружить другие кожные нарушения, например:
- Подмышечные веснушки
- Локализованные гипопигментации
- Ониходистрофии
- Гиперупругую кожу
Умственная отсталость, обычно легкой степени, наблюдается примерно у 30% пациентов. Около 25% пациентов имеют нейросенсорную потерю слуха. Судороги, нистагм или гипосмии были зарегистрированы у нескольких пациентов. Треть пациентов имеют невысокий рост, который станет очевидным, вскоре после рождения (большинство новорожденных имеют нормальный вес при рождении).
Несмотря на частое развитие пороков сердца, большинство пациентов остаются бессимптомными. Однако, в некоторых случаях, пациенты могут проявлять признаки патологий в сердце. Интересно, что более высокая частота семейной истории внезапной смерти и фибрилляции предсердий сообщалась у пациентов с гипертрофией левого желудочка без мутаций в гене PTPN11.
Около 35% пациентов демонстрируют различные черепно-лицевые аномалии. Глазной гипертелоризм является наиболее встречаемой лицевой аномалией (25%). Другие признаки включают:
- Нижнечелюстной прогнатизм
- Широкий носовой корень
- Дисморфозмы черепа
- Низкий посаженные уши
- Стоматологические аномалии
- Высокое небо
- Кожные складки
- Птоз
- Опухоли роговицы
Аномалии развития мочеполовой системы описаны у 26% больных, преимущественно у мужчин. Аномалии наружных половых органов, такие как крипторхизм или гипоспадия, можно обнаружить на медосмотре. Различные типы скелетных аномалий могут включать деформации грудной клетки, кифосколиоз, аномалии ребер, синдактилии, задержки развития или агенез постоянных зубов, сверхкомплектные зубы. Синдром LEOPARD может быть связан с гипертрофией сплетений, что может привести к невропатической боли.
Синдром LEOPARD. Диагностика
- КТ или МРТ головы
- Рентгенография скелета
- Эхокардиография
- Обследование мочеполовой системы
- УЗИ
- ЭКГ
Синдром LEOPARD. Лечение
Криохирургия и лазерное лечение могут быть полезными в лечении изолированных лентиго. Однако, из-за большого количества пятен лентиго, эти подходы могут быть очень трудоемкими. Для некоторых пациентов, лечение кремом третиноином и гидрохиноном может быть полезным.
Для лиц, больных структурными сердечными аномалиями, терапевтические схемы могут включать блокаторы бета-адренергических рецепторов или блокаторы кальциевых каналов.
Антиаритмические препараты могут потребоваться в случаях развития опасных для жизни желудочковых эктопий.
Хирургическое вмешательство может быть необходимым в случаях тяжелых деформаций и аномалий.
Синдром LEOPARD. Осложнения
Осложнения могут возникнуть из-за связанных аномалий.
Синдром LEOPARD. Прогноз
Прогноз в основном определяется по наличию сердечно-сосудистых осложнений. Большинство пациентов с синдромом LEOPARD могут вести нормальную жизнь. Сердечные патологии могут быть причиной смерти некоторых пациентов.
Источник
Синдром множественных пигментных пятен. Синдром LEOPARDТермин синдром «LEOPARD» является акронимом, состоящим из следующих элементов: lentigines — пигментные пятна; electrocardiographic defects — патология ЭКГ; ocular hypertelorism — глазной гипертело-ризм; pulmonary stenosis — стеноз легочной артерии; abnormalities of genitalia — патология гениталий; retardation of growth — низкий рост и sensorineural deafness — нейросенсорная глухота (Gorlin, Anderson, Blaw). Описанный случай был раньше подробно рассмотрен Gorlin, Anderson и Moller и Voron с сотр.. Клинические данные. Лицо. Обычно лицо имеет треугольную форму. Видны двусторонние теменные бугры, гипертелоризм, птоз, эпикант и часто крыловидные складки на шее. Покровная система. Когда имеются темно-коричневые пигментные пятна, внешний вид больных поразителен. Эти пятна многочисленны, малы (1—5мм) и от них свободны только поверхности слизистых оболочек. Они особенно обильны на шее и на туловище, по могут появляться па лице, волосистой части головы, ладонях и подошвах, а также па половых органах. Они могут быть видны уже при рождении, по могут появиться спустя короткое время после рождения. Число их быстро увеличивается с возрастом. Обычно по туловищу рассеяны несколько более крупных пятен темного цвета «черного кофе» (если мы можем создать новый термин). Пигментные пятна при синдроме «леопарда» отличаются от веснушек ранним выявлением и отсутствием связи с солнечным облучением, а микроскопически в них содержится большее число меланоцитов на единицу кожного покрова и выступающая сеть шиловидных клеток. Теперь ясно, что у многих больных может не быть пигментных пятен. В этих случаях идентификация синдрома зависит от наличия других характерных симптомов болезни и семейного их распространения. Сердечно-сосудистая система. Патология сердечно-сосудистой системы наиболее часто представлена клапанным стенозом легочной артерии, проявляющимся обычно в легкой степени. У одних больных наблюдается стеноз типичного клапанного типа, у других — необычные изменения клапанов легочной артерии, которые определяют термином «дисплазия клапанов легочной артерии» (Koretzky et аl.). Клапан легочной артерии состоит из трех отдельных створок, не соединенных между собой комиссурами. Закрытие отверстия обусловливается утолщением створок клапана легочной артерии неорганизованной слизистой тканью, которое приводит к их неподвижности. Клинически у больных с этим видом клапанной аномалии наблюдается выраженный систолический шум над легочной артерией, но без звука захлопывания клапана. Подобные же изменения могут более или менее часто наблюдаться в аортальных клапанах. У некоторых больных отмечается гипертрофия миокарда, развивающаяся как в результате субаортального, так и субпульмонального стеноза, охватывающая в первую очередь межелудочковую перегородку (Polani, Moynahan, Somerville, Bonham-Carter). В других работах описывается сочетанный дефект межпредсердной перегородки с воронкообразным или надклапанным стенозом легочной артерии или мышечным субаортальным стенозом (Korctzky et al., Lynch). При этом синдроме, не считающимся одним из типов порока сердца, отмечается характерная уникальная и постоянная аномалия ЭКГ. Эта аномалия лучше всего определяется средней осью комплекса QRS во фронтальной плоскости, обычно расположенной между —60° с —120° (S1, S2, S3 отведения). Эта патология может не быть демонстративной у каждого больного, но она может наблюдаться у других больных с синдромом, не имеющих структурной патологии сердца. У некоторых больных на ЭКГ был выявлен полный сердечный блок, полублок или полный блок одной из ножек пучка Гиса (Smith et al.).
Костно-мышечная система. Часто наблюдается задержка роста. У 85% больных оба показателя роста и веса ниже нормы на 25%, У большинства отмечается снижение обоих параметров на 10%. Постоянно наблюдаются: килевидпая или впалая грудь, кифоз в грудном отделе позвоночника, повышенное сгибание в пястно-фаланговых суставах пальцев, крыловидные лопатки. Мочеполовая система. Примерно у половины мужчин наблюдается гипоспадия (Gorlin et al.). Часто также отмечается крипторхизм или опущение только одного яичка. У некоторых женщин яичники отсутствуют или гипопластичпы. На основании опубликованных случаев и неопубликованного материала создается впечатление, что, так как большинство случаев передается через женщин, гипоплазия гонад выражена значительно более резко у мужчин. Часто наблюдается также позднее появление менструаций. Нервная система. Примерно у 20% больных выявлена легкая умственная отсталость (Moynahan, Watson, Vickers, Мас-Millan, Pickering et al.). Орган слуха. Примерно у 25% больных отмечается нейросенсорная глухота (Pickering et al.). Степень потери слуха у больных варьирует, по чаще она легкая. В противоположность этому у матери и дочери, описанных Capute с соавт., наблюдалась врожденная резко выраженная нейросенсорная глухота и очень плохо развитая речь). Из-за глубокой глухоты было невозможно произвести специальные аудиометрические пробы. Lassonde с сотр. отметили у своего больного глухонемоту. Вестибулярная система. Вестибулярные пробы были описаны Capute с соавт. Калорическая проба у 2 его больных патологии не выявила. Лабораторные данные. Электрокардиограммы. Наиболее частой патологией ЭКГ является дефект проводимости, хотя могут наблюдаться блок ножки пучка Гиса, аномальная волна, удлинение интервала Р — R, изменение комплекса ST и волны Т, а также расширение комплекса QRS (Matthews, Smith et al., Somerville, Bonham-Carter). – Также рекомендуем “Патология, наследственность и дифференциация синдрома LEOPARD” Оглавление темы “Наследственные болезни с глухотой”:
|
Источник