
Синдром патау по мкб 10

Содержание
- Описание
- Симптомы
- Диагностика
- Лечение
- Прогноз
Названия
Синдром Патау.

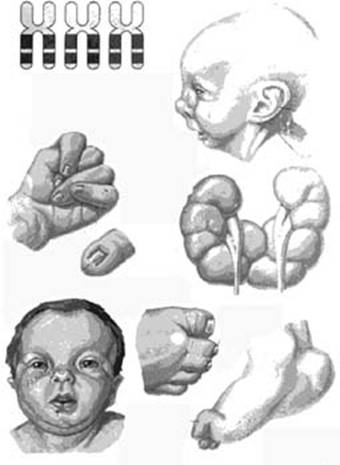
Внешний вид ребенка с синдромом Патау (трисомия 13)
Описание
Описан данный синдром в 1960 году, связан с нарушением 13-й хромосомы. Среди новорожденных синдром Патау встречается в соотношении 1:7000. Нет достоверных отличий по частоте встречаемости среди мужского и женского пола. Почти в 50% беременность осложняется многоводием. Масса тела новорожденного значительно ниже нормы при нормальной продолжительности; нередко в родах отмечается асфиксия.
Симптомы
Клиническая картина типична: микроцефальный череп с низким скошенным лбом, наблюдаются вдавленные височные области и дефекты кожи. Обращают на себя узкие глазные щели, расположены горизонтально, характерен глазной гипотелоризм. Отмечается глазная патология: микрофтальмия, колобомы, помутнение роговицы. Нос плоский, широкий, с запавшим переносьем и тупым втянутым кончиком. Низко расположенные ушные раковины сочетаются с маленькими мочками ушей, которые прижаты к голове. Завитки неправильной формы, козелок гипоплазирован. Неправильное развитие кортиева органа нередко обуславливает глухоту.
Одним из частых признаков синдрома является расщелина верхней губы и неба. Со стороны костно-мышечной системы имеют место полидактилия, флексорное положения кистей со своеобразным положением пальцев: 2-4 согнуты, приведены к ладони и перекрыты большим пальцем и мизинцем. Очень часто обнаруживаются пупочные и паховые грыжи, аномалии наружных половых органов: крипторхизм, гипоспадию, гипоплазию мошонки и полового члена у мальчиков, гипертрофию клитора и половых губ у девочек. У детей выявляют глубокую умственную отсталость, у них резко выражено моторное недоразвитие, нередко встречается судорожный синдром.
При патологоанатомическом исследовании обнаруживается множество дефектов развития практически всех систем органов. Масса мозга меньше нормы, недоразвиты или отсутствуют обонятельные луковицы и тракты, мозг часто не разделен на полушария, есть гипоплазия лобных долей и хиазмы зрительного нерва, мозжечка, агенезия мозолистого тела, гидроцефалия. Почти всегда выявляют аномалии сердца и сосудов: нередки дефект межжелудочковой и межпредсердной перегородок, диагностируется незаращение артериального протока, а также патология клапанного аппарата, стеноз легочной артерии. У половины больных с синдромом Патау регистрируют пороки развития почекмочевыводящих путей: кистозная почка, гидронефроз, дисплазия почек, удвоение почек и С такой же частотой встречаются различные дефекты и аномалии расположения органов пищеварения: аномалии поворота кишечника, патология брыжейки, дивертикул Меккеля, изменения поджелудочной железы в виде кистозно-фиброзного процесса. У 50% девочек находят удвоение влагалища и двурогую матку.
Дерматографические изменения при синдроме Патау (трисомия 13)
Диагностика
Дерматографические изменения заключаются в поперечной ладонной складке, дистальном расположении осевого трирадиуса, повышенной частоте радиальных петель и дуг в основном на большом пальце руки, диагностируется повышение частоты узоров на теноре.
Цитогенетическое исследование выявляет регулярную трисомию хромосомы 13 в 80% случаев. В остальных случаях отмечаются различные формы мозайцизма с участием трисомного клона, транслокации обычного типа t (13qDq). Описанные случаи частичной трисомии 13 чаще являются следствием перицентрической инверсии или сбалансированной транслокации у родителей.
Если при мозайцизме встречаются в основном те же фенотипические признаки, что и при «регулярной» трисомии, то проявления неполной трисомии хромосомы 13 отличаются от типчного синдрома Патау в зависимости от того, какой участок находится в трисомном положении.
Лечение
Лечебная помощь с синдромом Патау неспецифическая: по жизненным показаниям проводят операции по поводу врожденных пороков развития, необходимо общеукрепляющее лечение. Больные дети требуют тщательного ухода, следует проводить профилактику простудных и инфекционных болезней. Дети с синдромом Патау практически всегда имеют глубокую идиотию.
Прогноз
Прогноз для жизни при синдроме Патау неблагоприятный, средняя продолжительность жизни 130 дней: 60% всех больных умирают в первые 3 месяца после рождения, только 10% всех детей живут дольше года.
Источник
Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 1 мая 2019; проверки требуют 7 правок.
Синдром Пата́у (трисомия 13) — генетическое заболевание человека, которое характеризуется возникновением геномной мутации, а именно трисомией по 13-й хромосоме.
История
Трисомия 13 впервые описана Эразмусом Бартолином в 1657 году. Хромосомную природу заболевания выявил доктор Клаус Патау в 1960 году. Заболевание названо в его честь. Синдром Патау также был описан для племён с островов Тихого океана. Считается, что эти случаи были вызваны радиационным заражением, появившимся в результате испытаний ядерного оружия в регионе.
Этиология и эпидемиология
Встречается с частотой 1:7000-1:14000. Имеются два цитогенетических варианта синдрома Патау: простая трисомия и робертсоновская транслокация. Другие цитогенетические варианты (мозаицизм, изохромосома, неробертсоновские транслокации) обнаружены, но они встречаются крайне редко. Клиническая и патологоанатомическая картины простых трисомных форм и транслокационных не различается. 75 % случаев трисомии хромосомы 13 обусловлено появлением дополнительной хромосомы 13. Между частотой возникновения синдрома Патау и возрастом матери прослеживается зависимость, хотя и менее строгая, чем в случае синдрома Дауна. 25 % случаев СП — следствие транслокации с вовлечением хромосом 13-й пары, в том числе в трёх из четырёх таких случаев мутация de novo. В четверти случаев транслокация с вовлечением хромосом 13-й пары имеет наследственный характер с возвратным риском 14 %.
Соотношение полов при синдроме Патау близко к 1:1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25 — 30 % ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок беременности 38,5 недель). Риск возникновения этого синдрома у потомства увеличивается с возрастом матери, достигая пика в среднем к 31 году[2].
Проявления заболевания
Характерным осложнением беременности при вынашивании плода с синдромом Патау является многоводие: оно встречается почти в 50 % случаев Синдрома Патау.
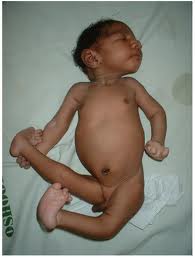
При синдроме Патау наблюдаются тяжёлые врождённые пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорождённых встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезёнки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития.
В связи с тяжёлыми врождёнными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95 % — до 1 года).
Однако некоторые больные живут в течение нескольких лет. Более того, в развитых странах отмечается тенденция увеличения продолжительности жизни больных синдромом Патау до 5 лет (около 15 % детей) и даже до 10 лет (2 — 3 % детей).
Оставшиеся в живых страдают глубокой идиотией.
Другие синдромы врождённых пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по отдельным признакам совпадают с синдромом Патау. Решающим фактором в диагностике является исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей.
Лечение
Исправить хромосомные нарушения невозможно. Комплексная работа группы различных специалистов заключается в постоянном контроле за состоянием здоровья больного и поддержке семьи.
См. также
- Хромосомные заболевания
- Синдром Дауна
- Синдром Эдвардса
Примечания
Ссылки
- https://web.archive.org/web/20070929094344/https://schools.keldysh.ru/school1413/pro_2005/z/hrbol.htm
- https://www.medkurs.ru/lecture2k/genetics/gl19/4288.html
- https://rh-conflict.narod.ru/student/lectures/hrombol.htm
Источник
Рубрика МКБ-10: Q91.7
МКБ-10 / Q00-Q99 КЛАСС XVII Врожденные аномалии пороки развития, деформации и хромосомные нарушения / Q90-Q99 Хромосомные аномалии, не классифицированные в других рубриках / Q91 Синдром Эдвардса и синдром Патау
Определение и общие сведения[править]
Трисомия по 13-й хромосоме
Синонимы: синдром Патау
Трисомия 13 представляет собой хромосомную аномалию, вызванную наличием дополнительной хромосомы 13 и характеризуется пороками развития головного мозга (голопроэнцефалия), лицевым дисморфизмом, глазными аномалиями, постсаксиальными полидактилиями, висцеральными мальформациями (кардиопатия) и тяжелой задержкой психомоторного развития.
Заболеваемость синдромом Патау оценивается в пределах от 1/8000 до 1/15 000 родов.
Этиология и патогенез[править]
Свободная трисомия 13 встречается примерно в 75% случаев. В 20% случаев трисомия 13 связана с транслокацией Робертсона, в которой сверхновая хромосома 13 присоединяется к другой акроцентрической хромосоме (хромосоме 13, 14, 15, 21 или 22). В редких случаях синдромом Патау вызван взаимной транслокацией между хромосомой 13 и неакроцентрической хромосомой. Мозаичная трисомия 13, при которой имеются как трисомные, так и нормальные клетки, сообщалась у нескольких пациентов с клинической картиной, которая варьировалась между нормальным фенотипом и классической трисомией 13 в зависимости от количества трисоматических клеток, присутствующих в тканях.
Клинические проявления[править]
Внутриутробная смертность при синдроме Патау составляет более 95%. Неврологические проявления тяжелые – с гипотонией и гипореактивностью, с очевидным отсутствием осознания окружающих. Голопрозэнцефалия (вызванная дефектом в разделении мозга на два полушария) присутствует в 70% случаев и может проявляться на МРТ в виде слияния полушарий различной степени. Аномалии лицевого черепа могут варьироваться от гипертелоризма и верхнечелюстного агенеза (80% случаев) до цебоцефалии или циклопии с отсутствием скелета носа. Также могут присутствовать расщепление губы/неба, микро- или анофтальмия, колобома (даже при отсутствии серьезных уродств головного мозга), участки затылочной кожной аплазии, постсаксиальная полидактилия, пороки развития сердца (80% случаев) и мочеполовой системы.
Синдром Патау неуточненный: Диагностика[править]
Трисомия 13 может быть заподозрена во время беременности на основнии УЗИ (голопрозэнцефалия, полидактилия) и может быть подтверждена кариотипическим анализом плода.
Дифференциальный диагноз[править]
Синдром Патау неуточненный: Лечение[править]
Лечение только поддерживающее.
Хирургическое лечение пороков развития мало способствует улучшению неблагоприятного прогноза для синдрома Патау: половина младенцев умирает в течение первого месяца жизни, а 90% умирают в возрасте до 1 года от сердечных, почечных или неврологических осложнений. Сообщалось о случаях хорошей выживаемости (в некоторых случаях во взрослую жизнь) в случаях мозаики или частичной трисомии, а также при отсутствии серьезных пороков развития головного мозга. В целом у немозаичных пациентов развивается только ограниченная автономия (отсутствие речи и способности к передвижению).
Профилактика[править]
Риск повторения трисомии (21, 13 или 18) в семьях со случаями синдрома Патау составляет около 1%. Однако в семьях, в которых трисомия 13 связана с транслокацией (робертсоновской или сбалансированной), риск повторения выше, если один из родителей является носителем сбалансированной транслокации.
Прочее[править]
Источники (ссылки)[править]
https://www.orpha.net
Дополнительная литература (рекомендуемая)[править]
Действующие вещества[править]
Источник
Медицинский эксперт статьи
х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Патау (трисомия по 13-й хромосоме) развивается при наличии лишней 13-й хромосомы и включает пороки развития переднего мозга, лица и глаз; выраженную умственную отсталость; низкую массу тела при рождении.
Синдром Патау (трисомия по 13-й хромосоме) встречается примерно в 1/10 000 живорождений; примерно в 80% имеет место полная трисомия 13. С возрастом матери увеличивается вероятность рождения ребенка с синдромом Патау, и лишняя хромосома, как правило, достается ребенку от матери.
Код по МКБ-10
Q91.7 Синдром Патау неуточненный
Что вызывает синдром Патау?
Дети с синдромом чаще рождаются у женщин старше 35 лет. Вероятность повторного рождения ребёнка с трисомией по хромосоме 13 в семьях не превышает 1% и не является противопоказанием для повторного деторождения.
Симптомы синдрома Патау
Дети рождаются маленькими к сроку гестации. Аномалии средней линии (например, дефекты кожи головы, кожных синусов) являются характерными. Голопрозенцефалия (нарушение нормального разделения переднего мозга) встречается часто. Сопутствующими аномалиями лица часто являются расщелины губы и нёба. Микрофтальмия, колобомы (расщелины радужки) и дисплазия сетчатки также встречаются часто. Надглазничные края не выражены, глазные щели часто узкие. Ушные раковины сформированы неправильно и обычно низко расположены. Часто у ребенка отмечается глухота. Рыхлые складки кожи нередко расположены на задней поверхности шеи. Также частыми являются обезьянья складка (единственная поперечная борозда на ладони), полидактилия, чрезмерно выпуклые узкие ногти. Примерно в 80 % случаев отмечаются тяжелые врожденные пороки развития сердечнососудистой системы; нередко встречается декстрокардия. Часто отмечаются аномалии половых органов как у мальчиков, так и у девочек; у мальчиков наблюдаются крипторхизм и аномалии мошонки, а у девочек характерно наличие двурогой матки. В первые месяцы жизни часто отмечаются эпизоды апноэ. Отмечается тяжелая умственная отсталость.
Синдром Патау характеризуется врождёнными дефектами глаз (колобома радужки, микрофтальм и анофтальм), незаращением верхней губы и/или нёба, аплазией кожи волосистой части головы, постаксиальной полидактилии, врождёнными пороками сердца и голопрозэнцефалией. Характерно наличие множественных врождённых пороков головного мозга и глубокой умственной отсталости. Высокая летальность – не более 10% детей доживают до 1 года.
Диагностика синдрома Патау
Диагноз можно предположить пренатально по нарушениям при УЗИ плода (например, задержка внутриутробного развития) или результатам скринингового обследования матери, а после рождения ребенка по его характерной внешности.
Подтверждение в обоих случаях проводится при исследовании кариотипа.
[1], [2], [3], [4], [5], [6], [7], [8], [9]
Какие анализы необходимы?
Какой прогноз имеет синдром Патау?
У большей части пациентов (80 %) синдром Патау сопровождается такими тяжелыми пороками развития, что они умирают на первом месяце жизни; менее 10 % живут более одного года.
Источник
Содержание
- Описание
- Симптомы
- Диагностика
- Лечение
Названия
Синдром Эдвардса.
Симптомы при синдроме Эдвардса (трисомия 18)
Описание
Впервые трисомию по группе Е описали J. Edwads и соавт. (1960г. ). Частота синдрома Эдвардса среди новорожденных в среднем составляет 1:7000, замечено, что девочки поражаются в 3 раза чаще, чем мальчики. Ученым высказано предположение о стабилизирующем действии Х-хромосомы при абберациях пары 18, тогда как зиготы с трисомией 18, имеющие мужской генотип, элиминируются. Средний возраст матерей 32,5 года, отцов – 35 лет.
Симптомы
Длительность беременности превышает нормальную, и составляет в среднем 42 недели. Во время беременности диагностируют слабую активность плода и многоводие. Отмечается,что плацента имеет меньшие, чем обычно, размеры, часто обнаруживается только одна пупочная артерия; многие дети рождаются в асфиксии, с очень низкой массой тела и выраженной гипотрофией.
Фенотип больных с синдромом Эдвардса довольно характерен. Череп долихоцефалический, с низким лбом, затылок более широкий и выступающий. Часто встречается микроцефалия или гидроцефалия. Надглазничные валики сглажены, глазные щели узкие, наблюдается эпикант, птоз, часто встречается глазная патология, микрофтальмия, колобома, катаракта. Переносье вдавлено, но спинка носа тонкая, выступающая, ушные раковины расположены очень низко, диагностируется отсутствие мочки и козелка, недоразвиты завиток и противозавиток. Характерна микроретрогнатия. Рот мешьше обычных размеров, имеет треугольную форму, верхняя губа короче обычного. Небо высокое, иногда с расщелиной, шея короткая, нередко с крыловидной складкой.
Аномалии опорно-двигательного аппарата отличаются разнообразием: расширение шрудной клетки, укорочение грудины, таз узкий, деформации конечностей, ограничена подвижность в тазобедренных суставах, описаны вывихи бедра. Кисти и пальцы короткие, клинодактилия 5 пальцев кисти, дистально расположенный, гипоплазированный 1 палец кисти, сглажен тенар. Пальцы сжаты в кулак по типу «флексорной аномалии»: 2 и 5 пальцы расположены сверху и прикрывают прижатые к ладони 3 и 4 пальцы; 1 палец стопы широкий и короткий, синдактилия 2 и 3 пальцев. Типична для трисомии 18 форма стопы в виде своеобразной «качалки». Характерна общая мышечная гипотония. У мальчиков нередко встречается крипторхизм, гипоспадия; гипертрофия клитора у девочек.
Интеллектуальный дефект соответствует олигофрении в степени идиотии ил глубокой имбецильности и только в единичных описаниях мозаичного варианта трисомии 18 умственное недоразвитие менее грубое. Нередко у больного развиваются судороги.
Внешний вид больного с синдромом Эдвардса (трисомия 18)
Диагностика
Дерматографическая картина при синдроме Эдвардса отличается своеобразными признаками: отмечается большая частота дуг на подушечках пальцев рук (примерно в 10 раз выше, чем в общей популяции), нередко дистальная сгибательная складка на пальцах не развита, у трети больных обнаруживается поперечная ладонная борозда, гребневый счет увеличен, осевой трирадиус обычно расположен дистально.
При аутопсии при синдроме Эдвардса находят разнообразные пороки развития практически всех органов и систем. С различной частотой встречаются аномалии ЦНС: аплазия или гипоплазия мозолистого тела, гипоплазия мозжечка, атрофия мозговых извилин. У 95% пациентов с трисомией 18 диагностируются пороки сердца и крупных сосудов, наиболее часто встречаются дефект межжелудочковой перегородки и незаращение артериального протока. Около половины всех случаев синдрома Эдвардса сопровождается аномалиями органов пищеварения: нарушение расположения кишечника, меккелев дивертикул, атрезия пищевода, атрезия заднего прохода. В 50% случаев отмечаются пороки развития мочеполовой системы – дольчатая или подковообразная почка, дупликация мочеточников, гипоплазия яичников.
При цитогенетическом обследовании в 80% случаев находят трисомию 18, у 10% больных – мозайцизм. Описаны случаи транслокационного варианта, двойной анеуплодии типа 48, ХХУ +18 с участием трисомного по хромосоме 18 клона.
Лечение
Нет патогенетического лечения синдрома Эдвардса на сегодняшний день, возможна лишь симптоматическая коррекция патологий. Прогноз для жизни неблагоприятный, средняя продолжительность жизни мальчиков 2-3 месяца, девочек – 10 месяцев. 30% больных умирают в течение 1-го года жизни, до года доживает лишь 10% больных. При мозаичных вариантах прогноз для жизни несколько лучше.
Источник