
Синдром птицеголовая карликовость или синдром секкеля

Синдром Секкеля является унаследованной формой врожденной карликовости , а это значит, что младенец начинает очень мало и не может нормально расти после рождения. В то время как люди с синдромом Секкеля обычно будут пропорциональны по масштабу, у них будет отчетливо малый размер головы. Также распространена умственная отсталость.
Несмотря на множество физических и умственных проблем, с которыми сталкивается человек с синдромом Секкеля, многие из них, как известно, хорошо живут более 50 лет.
Причины синдрома Секкеля
Синдром Секкеля является наследственным расстройством, связанным с генетическими мутациями на одной из трех разных хромосом. Считается, что это бывает крайне редко, с немногим более 100 случаев, зарегистрированных с 1960 года. Многие дети, получившие диагноз синдрома Секкеля, родились у родителей, которые тесно связаны (родственники), например, брак с кузенами или братьями и сестрами.
Синдром Секкеля является рецессивным генетическим заболеванием, он возникает только тогда, когда ребенок наследует один и тот же ненормальный ген от каждого родителя. Если ребенок получает один нормальный ген и один ненормальный ген, ребенок будет носителем синдрома, но обычно не проявляет симптомов.
Если оба родителя имеют одинаковую хромосомную мутацию для синдрома Секкеля, их риск иметь ребенка с синдромом Секкеля составляет 25%, а риск наличия носителя — 50%.
Характеристики синдрома Секкеля
Синдром Секкеля характеризуется аномально медленным развитием плода и низким весом при рождении.
После рождения у ребенка будет медленный рост и созревание костей, что приводит к короткому, но пропорциональному росту (в отличие от ахондроплазии). Лица с синдромом Секкеля обладают различными физическими и характеристиками развития, в том числе:
Очень маленький размер и вес при рождении
- Чрезвычайно малый, пропорциональный рост
- Аномально небольшой размер головы (микроцефалия)
- Клювообразный выступ носа
- Узкое лицо
- Нарушенные уши
- Необычно маленькая челюсть (микрогнатия)
- Умственная отсталость, часто тяжелая с IQ менее 50
Другие симптомы могут включать аномально большие глаза, высокое арочное небо, повреждение зубов и другие деформации кости. Расстройства крови, такие как анемия (низкие эритроциты), панцитопения (недостаточно клеток крови) или острый миелоидный лейкоз.
В некоторых случаях семенники у мужчин не спускаются в мошонку, а женщины могут иметь аномально увеличенный клитор. Кроме того, у людей с синдромом Секкеля могут быть чрезмерные волосы на теле и единая глубокая складка на ладонях (известная как складки обезьян).
Диагностика синдрома Секкеля
Диагноз синдрома Секкеля основан почти исключительно на физических симптомах. Возможно, потребуется рентгеновское изображение и другие инструменты (МРТ, КТ), чтобы отличить его от других подобных условий. В настоящее время нет лабораторных или генетических тестов, специфичных для синдрома Секкеля. В некоторых случаях окончательный диагноз не может быть сделан до тех пор, пока ребенок не станет старше и не появятся характерные симптомы.
Лечение синдрома Секкеля
Лечение синдрома Секкеля сосредоточено на любой медицинской проблеме, которая может возникнуть, особенно при нарушениях крови и структурных деформациях. У людей с психическими расстройствами и их семьям должна быть предоставлена соответствующая социальная поддержка и консультационные услуги.
Источник
Что такое синдром Секкеля?
Синдром Секкеля чрезвычайно редкое наследственное заболевание, характеризующееся задержкой роста до рождения (задержка внутриутробного развития), что приводит к низкой массе тела при рождении. Задержка роста продолжается после рождения (послеродовой период), что приводит к низкорослости (карликовость). Другие симптомы и физические особенности, связанные с синдромом Секкеля, включают аномально маленькую голову (микроцефалия); разную степень умственной отсталости; и/или необычные характерные черты лица, включая «клювоподобный» выступ носа. Другие черты лица могут включать аномально большие глаза, узкое лицо, деформированные уши и/или необычно маленькую челюсть (микрогнатия). Кроме того, у некоторых младенцев может наблюдаться постоянная фиксация пятых пальцев в согнутом положении (клинодактилия), деформация (дисплазия) тазобедренного сустава, вывих кости в предплечье (радиальный смещение) и/или другие физические отклонения.
Признаки и симптомы
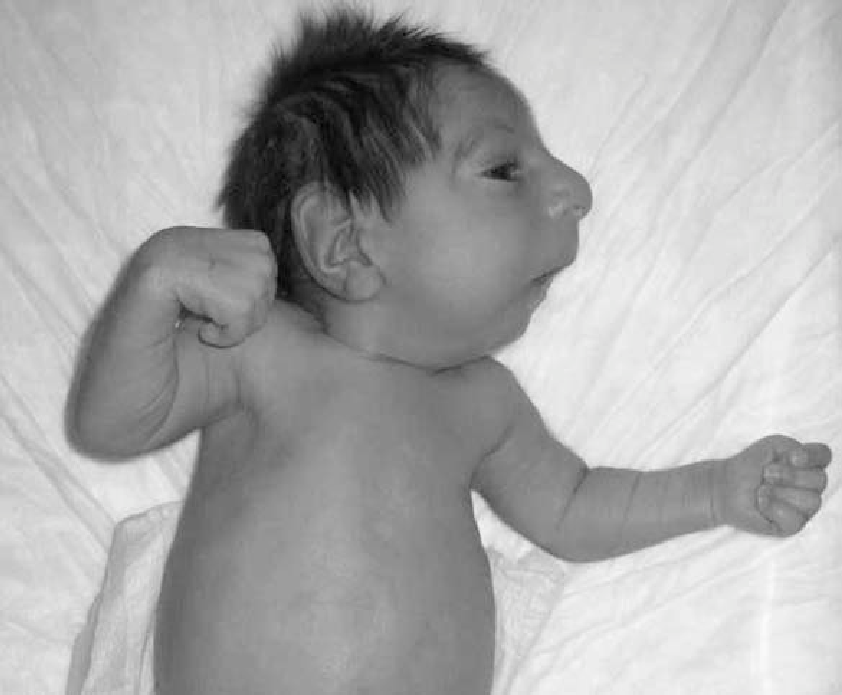
Синдром Секкеля характеризуется аномально медленным развитием плода и низкой массой тела при рождении. После рождения у ребенка будет наблюдаться медленный рост и созревание костей, что приведет к низкому, но пропорциональному росту (в отличие от карликовости коротких конечностей или ахондроплазии). Больные с синдромом Секкеля имеют различные физические отклонения и особенности развития, в том числе:
- Очень маленький размер и вес при рождении (в среднем 3,3 фунта или 1,4 кг);
- Чрезвычайно маленький, пропорциональный рост;
- Аномально маленький размер головы (микроцефалия);
- Клювовидный выступ носа;
- Узкое лицо;
- Деформированные уши;
- Необычно маленькая челюсть (микрогнатия);
- Умственная отсталость, часто тяжелая, с IQ (коэффициент интеллекта) менее 50.
Другие симптомы могут включать аномально большие глаза, высокое арочное небо, пороки развития зубов и другие деформации костей. Также часто наблюдаются такие заболевания крови, как анемия (низкий уровень эритроцитов), панцитопения (недостаток клеток крови) или острый миелоидный лейкоз (тип рака крови).
В некоторых случаях у мужчин яички не могут опускаться в мошонку (крипторхизм), а у женщин может быть ненормально увеличенный клитор. Кроме того, у больных с синдромом может быть чрезмерное оволосение на теле и одна единственная глубокая складка на ладонях (известная как обезьянья складка).
Причины
Синдром Секкеля — чрезвычайно редкое заболевание, передающееся по аутосомно-рецессивному типу. Три варианта синдрома Секкеля включают нарушения или изменения (мутации) генов на трех разных хромосомах. Расположение генной карты: синдром Секеля 1 на хромосоме 3 (3q22-q24); Синдром Секкеля 2 на хромосоме 18 (18p11.31-q11) и синдром Секеля 3 на хромосоме 14 (14q21-q22).
Хромосомы, присутствующие в ядре клеток человека, несут генетическую информацию для каждого человека. Клетки человеческого тела обычно имеют 46 хромосом. Пары человеческих хромосом пронумерованы от 1 до 22, а половые хромосомы обозначены X и Y. У мужчин одна X и одна Y хромосома, а у женщин две X хромосомы. У каждой хромосомы есть короткое плечо, обозначенное буквой «p», и длинное плечо, обозначенное буквой «q». Хромосомы далее подразделяются на множество пронумерованных полос. Например, «хромосома 3q22-q24» относится к области между полосой 22 и полосой 24 на длинном плече хромосомы 3. Подобным образом «хромосома 18p11.31-q11.2» относится к более широкой области между полосой 11p31 на коротком плече хромосомы 18 и полосе 11.2 на длинном плече хромосомы 18. Опять же, хромосома 14q21-q22 относится к более узкой области на длинном отрезке хромосомы 14 между полосами 21 и 22. Пронумерованные полосы указывают расположение тысяч генов, присутствующих на каждой хромосоме.
Специфический ген, участвующий в синдроме Секкеля 1, известен как ген атаксии-телеангиэктазии и ген Rad3-родственного белка (ATR). Гены, участвующие в синдроме Секкеля 2 и 3 типов, неизвестны.
Генетические заболевания определяются комбинацией генов определенного признака, которые находятся на хромосомах, полученных от отца и матери.
Рецессивные генетические расстройства возникают, когда человек наследует один и тот же аномальный ген одного и того же признака от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, он будет носителем болезни, но обычно бессимптомным. Риск для двух родителей-носителей передать дефектный ген и, следовательно, иметь больного ребенка, составляет 25% при каждой беременности. Риск иметь ребенка, который будет носителем, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей и будет генетически нормальным по данному признаку, составляет 25%. Риск одинаков для мужчин и женщин.
Все люди несут несколько аномальных генов. Родители, которые являются близкими родственниками (кровными родственниками), имеют больше шансов, чем неродственные родители, иметь один и тот же аномальный ген, что увеличивает риск рождения детей с рецессивным генетическим заболеванием.
Затронутые группы населения
Синдром Секкеля — чрезвычайно редкое наследственное заболевание, которое поражает в равной степени мужчин и женщин. Точная частота расстройства неизвестна. С момента первоначального описания в 1960 г. в медицинской литературе было зарегистрировано более 100 случаев.
Близкие по симптомам расстройства
Симптомы следующих расстройств могут быть похожи на симптомы синдрома Секкеля. Сравнения могут быть полезны для дифференциальной диагностики:
Синдром Секкеля — один из шести расстройств в классе карликовости, известном как «изначальная карликовость». Эти расстройства имеют схожие характеристики, включая пороки развития скелета (дисплазию), дефицит роста до рождения (задержка внутриутробного развития) и в младенчестве и детстве, что в конечном итоге приводит к разной степени низкого роста. В эту группу заболеваний в настоящее время входят пять расстройств: синдром Секкеля, синдром Мейера-Горлина; Синдром Рассела-Сильвера; Остеодиспластическая примордиальная карликовость Маевского I/III; и остеодиспластическая примордиальная карликовость Маевского II типа.
Остеодиспластическая примордиальная карликовость Маевского II типа является чрезвычайно редким генетическим заболеванием, характеризующимся низким ростом, низкой массой тела при рождении, аномально маленькой головой (микроцефалия) и/или аномалиями скелета. Другие физические признаки могут включать большие глаза, выступающий нос в форме клюва, опущенную челюсть и/или узкое лицо. Обычно возникает серьезная недостаточность роста до рождения (внутриутробная недостаточность роста). В некоторых случаях может присутствовать умственная отсталость. Также могут появиться различные дополнительные симптомы. Конкретные симптомы и степень тяжести варьируются от случая к случаю. Считается, что II тип остеодиспластической примордиальной карликовости Маевского наследуется по аутосомно-рецессивному типу.
Диагностика
С появлением технически совершенной ультасонографии синдром Секкеля можно диагностировать до рождения (пренатально). При ультразвуковом исследовании плода используются отраженные звуковых волны для создания изображения развивающегося плода. После рождения можно заподозрить синдром Секкеля на основании тщательного клинического обследования, подробного анамнеза пациента и различных специализированных тестов. Хотя характерные черепно-лицевые, скелетные и/или другие физические аномалии, связанные с синдромом Секкеля, могут присутствовать при рождении, в некоторых случаях диагноз может не подтверждаться до тех пор, пока больной ребенок не вырастет и не разовьется полный синдром (например, когда станет очевидным низкий рост и/или умственная отсталость).
Низкий рост, связанный с синдромом Секкеля, предполагает пропорциональный рост рук и ног, что позволяет проводить дифференциальную диагностику от синдромов, связанных с низким ростом и аномально маленькими руками и ногами (карликовость с короткими конечностями).
Стандартные методы лечения
От синдрома Секкеля нет лекарства. Некоторые лекарства могут быть назначены для устранения других симптомов, связанных с заболеванием.
Лечение направлено на решение любой медицинской проблемы, которая может возникнуть, особенно заболеваний крови и структурных деформаций. Людям с умственными недостатками и их семьям потребуется соответствующая социальная поддержка и консультационные услуги.
Прогноз
Дети, страдающие синдромом Секкеля, могут жить в течение длительного периода времени, хотя они часто сталкиваются с глубокими психическими и физическими отклонениями.
Источник
Седых А.В. 1
1Тюменский государственный медицинский университет Министерства здравоохранения Российской федерации, Тюмень, Россия
Текст работы размещён без изображений и формул.
Полная версия работы доступна во вкладке “Файлы работы” в формате PDF
Введение
Актуальность. Низкорослость (нанизм) – достаточно часто встречающийся симптом различных наследственных, врождённых и приобретённых заболеваний детского возраста. Одним из примеров таких заболеваний является синдром Секкеля (сочетание карликовости с «птицеголовостью», микроцефалическая примордиальная карликовость I типа), в основе которого лежат мутации различных генов.
Цель: изучить развитие болезниСеккеля, ее этиологию, частоту встречаемости и
патогенез, а также лечение.
Задачи:
1) Проанализировать этиологию и встречаемость болезни
2) Изучить патогенез и клиническую картину болезни
3) Рассмотреть лечение болезни
Этиология и встречаемость
Впервые синдром описали Тревор Манн и Александр Рассел в 1959 году. Гельмут Секкель в 1960 году опубликовал статью, в которой привёл результаты обследования двух собственных пациентов и литературные данные о 24 больных, имевших сходные клинические проявления, а термины «птицеголовая карликовость» и «наноцефалия» принадлежат Рудольфу Вирхову.
Частота встречаемости в популяции составляет 1:10 000 случаев. Согласно генетическим данным, заболевание наследуется по аутосомно-рецессивному типу.(Описано 9 форм заболевания)
Развитие синдрома Секкеля 1 связывают с мутацией в гене ATR, локализованном на хромосоме 3q23. Ген является важнейшим регулятором геномной целостности, контролирует и координирует ДНК-репликацию. Поломки в гене ATR приводят к нарушению клеточного цикла и появлению нестабильности хрупких хромосомных участков, что связывают с развитием фенотипа синдрома Секкеля 1. Мутации в гене АТR отвечают также за развитие атаксии-телеангиэктазии и семейных форм рака.
Дети с синдромом Секкеля рождаются с выраженной внутриутробной гипоплазией: средняя масса тела доношенных новорождённых составляет 1600-1700 г, длина тела – 40-45 см. В последующие годы сохраняется пропорциональная низкорослость, чаще значительная, масса тела соответствует росту.
Патогенез
Наиболее характерными внешними признаками заболевания являются черепно-лицевые дизморфии, давшие основание для термина «птицеголовость»: микроцефалия с преждевременным закрытием черепных швов (краниосиностоз); задержка увеличения окружности головы, в поло- вине случаев соответствующая общей задержке роста, а в половине – даже превосходящая ее; скошенный лоб; выступающий загнутый («клювовидный») нос с утолщенным кончиком; микро- и ретрогения; низкорасположенные деформированные ушные раковины; относительно большие глаза с антимонголоидным разрезом; редкие волосы . Отмечаются также страбизм, аномалии зубов и ротовой полости (узкое высокое нёбо, его расщелины, частичная адонтия, гипоплазия эмали). Синдром может сопровождаться появлением гипопигментированных пятен на различных участках тела – наиболее часто они наблюдаются в области подмышечных впадин и на шее.
Диагностика
Диагностика заболевания основывается на клинических проявлениях. В пренатальной диагностике основную роль играет ультразвуковое исследование, которое позволяет выявить гипоплазию плода, микроцефалию, аномалии развития конечностей, черепно-лицевые аномалии. При данном синдроме возможно перенашивание беременности.
Лечение
При синдроме Секкеля лечение симптоматическое. Применяются препараты рекомбинантного гормона роста (рГР), препараты, улучшающие обменные процессы, ноотропы . Несмотря на выраженную задержку психического развития, раннее двигательное развитие может соответствовать возрасту. Больные обычно дружелюбны и общительны, но в то же время невнимательны и неусидчивы. Известно об одном больном синдромом Секкеля, дожившем до 75 лет.
Список литературы
Роберт Л. Ньюссбаум, Родерик Р. Мак-Иннес, Хантингтон Ф. Виллард «Медицинская генетика» (с.485-486)
https://simptomy-lechenie.net/ – Синдром Секкеля
Логачев М.Ф. Задержка внутриутробного развития и постнатальная низкорослость у детей: (Этиология, патогенез, клиническая картина, диагноз): дис. . д-ра мед. наук / Логачев М.Ф. М., 2000. – 289 с.
Источник
Синдром секкеля является врожденным заболеванием, характеризующимся наличием карликовости и задержки внутриутробного развития, которая продолжается до послеродовой стадии (Baquero Álvarez, Tobón Restrepo и Alzate Gómez, 2014).
На этиологическом уровне синдром Секкеля имеет аутосомно-генетическое происхождение рецессивного характера, связанное с различными специфическими мутациями и различными вариантами патологии, такими как локализация хромосомы 3, хромосомы 18 или 14 (Национальная организация редких заболеваний). , 2007).
С другой стороны, на клиническом уровне синдром Зеккеля отличается развитием микроцефалии, микогнатии, невысокого роста или особого внешнего вида лица (профиль птицы). Кроме того, все эти черты часто сопровождаются серьезной задержкой умственного развития..
Что касается диагноза этой патологии, его можно подтвердить во время беременности, поскольку морфологические признаки и патология, связанные с внутриутробным ростом, могут быть выявлены с помощью обычных ультразвуковых исследований (Luna-Domínguez, Iglesias-Leboreiro, Bernárdez-Zapata и Rendón -Macias, 2011).
В настоящее время нет лекарства от синдрома Зеккеля, лечение обычно направлено на генетическое исследование и лечение медицинских осложнений с помощью междисциплинарного подхода (Baquero Álvarez, Tobón Restrepo и Alzate Gómez, 2014).
Характеристика синдрома Секкеля
Синдром Секкеля – это редкое или редкое заболевание. Он характеризуется патологической задержкой роста плода во время беременности, которая приводит к развитию уменьшенного размера тела, микроцефалии, умственной отсталости или характерного внешнего вида лица, называемого профилем головы или птицы (Sanske et al., 1997, Bocchini, 2014).
Из-за своей низкой распространенности синдром Секкеля классифицируется как одно из редких заболеваний или расстройств, то есть тех, которые поражают очень небольшую группу людей в общей популяции по сравнению с другими типами патологий (Richter et al. , 2015).
Хотя существуют различные диапазоны распространенности, в случае Европы расстройство является частью редких заболеваний, когда оно встречается менее чем в одном случае на 2000 человек (Испанская федерация редких заболеваний, 2016 г.).
Как правило, редкие заболевания являются продуктом изменений или генетических мутаций, как в случае с синдромом Секкеля (Richter et al., 2015). Таким образом, эта патология была первоначально описана Рудольфом Вирховым в 1892 году, на основании своих медицинских данных он дал ей название «карликовость головы птицы».
Однако только в 1960 году Хельмонт Секкель описал окончательные клинические характеристики синдрома (Baquero Álvarez, TobónRestrepo and Alzate Gómez, 2014).
статистика
Как мы уже указывали, частота синдрома Секкеля недостаточна, поэтому в 2010 году в медицинской литературе было зарегистрировано около 100 случаев, среди которых было выявлено более 12 пострадавших семей (Бакеро Альварес, Тобон Рестрепо и Альзате Гомес). , 2014).
На определенном уровне различные эпидемиологические исследования оценили их частоту менее чем в 1 случае на 10 000 живорожденных детей. С другой стороны, синдром Секкеля – это патология, которая в равной степени затрагивает оба пола и не связана с каким-либо конкретным географическим регионом или этнической группой (Луна-Домингес, Иглесиас-Леборейро, Бернардес-Сапата и Рендон-Масиас, 2011).
Признаки и симптомы
Клинические признаки синдрома Зеккеля могут варьироваться в зависимости от степени их влияния, поскольку они будут зависеть в основном от их специфического этиологического происхождения..
Тем не менее, некоторые из наиболее частых признаков и симптомов этой патологии включают в себя (Faivre and Comier-Daire, 2005, Национальная организация по редким заболеваниям, 2007):
Задержка внутриутробного развития
Центральная медицинская находка этой патологии – наличие аномально медленного развития роста плода на стадии беременности..
Как мы уже указывали ранее, синдром Зеккеля включен в патологии, классифицируемые как карликовые, у которых есть существенная задержка роста и возраста кости, в основном.
Обычно медленное физическое развитие обычно распространяется после рождения, на неонатальной и младенческой стадии, и, как следствие, могут развиваться вторичные медицинские осложнения, такие как описанные ниже..
микроцефалия
Микроцефалия – это тип неврологической патологии, при котором фундаментальным клиническим открытием является наличие аномально уменьшенной окружности черепа, то есть размер головы больного человека меньше, чем ожидается для его пола и возрастной группы.
Микроцефалия может появиться в результате плохого развития черепных структур или из-за нарушения ритма роста.
Однако в случае синдрома Секкеля микроцефалия является продуктом задержки внутриутробного развития, поэтому череп и мозг плода не растут с постоянной скоростью и в соответствии с ожидаемым.
Хотя степень тяжести медицинских последствий микроцефалии варьируется, в целом она обычно сопровождается значительными задержками в развитии, дефицитом обучения, физическими недостатками, судорожными приступами и другими..
Кроме того, черепно-лицевая структура людей, пораженных синдромом Зеккеля, обычно имеет и другие особенности, такие как краниосиностоз, то есть раннее закрытие черепных швов..
Низкий рост
Другой важной особенностью синдрома Секкеля является наличие невысокого роста, в некоторых случаях в медицинской литературе называемого карликовостью..
Задержка внутриутробного развития приводит к низкой массе тела при рождении, что сопровождается задержкой развития или созревания кости..
Таким образом, во время постнатальной фазы эти характеристики приводят к развитию аномально уменьшенной высоты и конечностей..
Кроме того, это может также привести к развитию других типов патологий скелета, таких как лучевая дислокация, дисплазия тазобедренного сустава, кифосколиоз, клинофактерия или косолапость..
Профиль птицы
Черепно-лицевые изменения дают людям, страдающим синдромом Зеккеля, отличительную конфигурацию, характеризующуюся различными морфологическими признаками:
– микроцефалияУменьшенная окружность мозга, то есть ненормально маленькая голова.
– Уменьшенная фация: уменьшенное или ненормально маленькое расширение лица, обычно визуально воспринимаемое как удлиненное и узкое.
– Передняя известностьЛоб имеет видную или выступающую структурную конфигурацию.
– Выдающийся носовой мост: нос обычно имеет выступающую структуру в форме клюва, во многих случаях называемый пико-корнообразным носом.
– micrognatia: морфологические структуры челюсти имеют тенденцию быть меньше или меньше, чем обычно, что может вызвать важные изменения в питании.
– Большие глаза: по сравнению с остальными структурами глаза могут быть видны больше, чем обычно. Кроме того, в некоторых случаях можно наблюдать развитие измененных процессов, таких как экзофтальм или проптоз, то есть обилие глазных яблок..
– косоглазие: в некоторых случаях также можно наблюдать отклонение одного или обоих глазных яблок, они могут быть повернуты наружу или к носовой структуре.
– Диспластические ушиУши обычно имеют неполное или недостаточное развитие с отсутствием долей. Кроме того, они обычно имеют низкую черепно-лицевую имплантацию.
– Волчья пастьНа небе пораженные лица обычно имеют различные изменения, такие как арочная крыша или наличие трещин или трещин.
– Дисплазия зубовСтоматологические материалы также часто плохо развиты, плохо организованы и переполнены.
Дефицит интеллектуального развития
Недостаточное развитие черепно-мозговой структуры может вызвать серьезные неврологические и когнитивные нарушения у людей, страдающих синдромом Секкеля.
Таким образом, одним из наиболее частых результатов является наличие дефицита интеллектуального развития, характеризующегося плохой успеваемостью в языковой области, памяти, внимании и т. Д..
Кроме того, склонны появляться различные поведенческие и моторные изменения, такие как стереотипы или эпизоды агрессивности..
Другие особенности
В дополнение к признакам, указанным выше, в клиническом течении синдрома Секкеля могут появляться другие виды медицинских осложнений:
– Дисплазия половых органов: в случае пораженных мужчин часто встречается криптокида или недостаточное опускание яичек к мошонке. У женщин часто наблюдается клиторомегалия или аномально большой клитор..
– гирсутизм: этот термин обычно используется для обозначения чрезмерного или чрезмерного присутствия волос на поверхности тела.
– Гематологический дефицит: во многих случаях можно выявить значительный дефицит одного или нескольких компонентов крови (эритроцитов, лейкоцитов, тромбоцитов и т. д.).
причины
Синдром Секкеля – это патология с аутосомно-генетическим происхождением рецессивного характера, то есть необходимо наличие двух копий дефектного или измененного гена, чтобы могло развиться расстройство и его клинические характеристики (Faivre and Comier-Daire, 2005)..
Кроме того, с точки зрения специфических генетических аномалий синдром Зеккеля является широко гетерогенным, поскольку было выявлено до 3 типов изменений (Fitzgerald, O’Driscoll, Chong, Keating and Shannon, 2012), в частности, расположенных на хромосомах 3, 18 и 14 (Faivre yComier-Daire, 2005).
Кроме того, были выявлены три различные клинические формы синдрома Секкеля, связанные с генетическими изменениями (Faivre and Comier-Daire, 2005, Faivre and Comier-Daire, 2005):
– Синдром Секкеля 1: связан с изменениями в хромосоме 3, особенно в месте расположения 3q22-P24 и связан со специфической мутацией в гене белка Rad3.
– Синдром Секкеля 2: связан с изменениями в 18 хромосоме, в частности в месте 18p11.31-q11, однако специфическая мутация еще не идентифицирована.
– Синдром Секкеля 3: связан с изменениями в хромосоме 14, особенно в месте 14q21-q22, однако специфическая мутация еще не идентифицирована.
Однако другие исследования показывают, что синдром Секкеля может появиться в результате определенных генетических мутаций в следующих местах:
– Ген Rbbp8 на 18 хромосоме.
– Ген CNPJ на 13 хромосоме.
– Ген CEP152 на хромосоме 15.
– Ген CEP63 на хромосоме 3.
– Ген NIN на хромосоме 14.
– Ген ДНК2 на хромосоме 10.
– Ген TRAIP на хромосоме 3.
диагностика
Клинические и морфологические особенности синдрома Секкеля, такие как задержка внутриутробного развития, микроцефалия или структурные аномалии лица, могут быть выявлены во время беременности..
Таким образом, УЗИ плода являются одним из наиболее эффективных методов, они позволяют визуально и метрически обнаруживать структурные аномалии скелета и изменять ритмы физического развития (Национальная организация по редким заболеваниям, 2007)..
Тем не менее, этот тип патологии не может быть подтвержден клинически, пока медицинская картина полностью не сформирована, обычно в раннем детстве (Национальная организация по редким заболеваниям, 2007).
Кроме того, еще одним важным моментом является генетическое исследование, поскольку оно позволяет изучать историю семьи и наследственные паттерны..
лечение
В настоящее время не было выявлено какого-либо медицинского подхода для лечения или остановки прогрессирования синдрома Секкеля. Однако для улучшения симптомов можно использовать различные методы лечения (Baquero Álvarez, Tobón Restrepo и Alzate Gómez, 2014).
Таким образом, лечение обычно ориентировано на генетическое изучение и лечение медицинских осложнений с помощью междисциплинарного подхода (Baquero Álvarez, Tobón Restrepo and Alzate Gómez, 2014).
Кроме того, фундаментальным является контроль гематологических нарушений и, следовательно, лечение других вторичных медицинских осложнений, таких как анемия, панцитопения или лейкоз..
ссылки
- Baquero Álvarez, J., Tobón Restrepo, J., & Alzate Gómez, D. (2014). Два случая с синдромом Секкеля в колумбийской семье. Преподобный Мекс Педр, 69-73.
- Боккини, C. (2014). SECKEL СИНДРОМ. Получено из Университета Джона Хопкинса.
- Comier-Daire, V. & Faivre-Olivier. (2005). Синдром Секкеля Получено от Orphanet.
- Фицджеральд Б., О’Дрисколл М., Чонг К., Китинг С. и Шеннон П. (2012). Нейропатология эмбриональной стадии синдрома Секкеля: история болезни с морфологическим коррелятом для возникающих молекулярных механизмов Brain & Development, 238-243.
- Luna-Domínguez, C., Хосе Иглесиас-Леборейро, J., Бернардес-Сапата, I., и Rendón-Macías, M. (s.f.). Случай с синдромом Сиккела. Преподобный Мекс Педр.
- NORD. (2007). Синдром Секкеля Получено от Национальной организации по редким заболеваниям.
Источник