
Синдром робена наследуется двумя путями


Синдром Пьера Робена — внутриутробно сформированная аномалия, характеризующаяся патологическим развитием челюстно-лицевой области. В официальной медицине синдром также именуется аномалия Робена. Он имеет код по МКБ-10 Q87.0 и относится к врожденным аномалиям, изменяющим внешний вид лица и приводящим к уродству.
Заболевание было открыто в начале 20 века стоматологом из Франции П. Робеном. Он впервые описал клинические признаки синдрома и характерный внешний вид больных. У его пациента была очень мелкая голова, срезанный подбородок, аномальная структура языка. Больной ребенок с трудом мог дышать, жевать и глотать. Он отставал в психомоторном развитии от своих сверстников.
Этот врожденный дефект встречается достаточно редко: у 1 из 30000 живорожденных, одинаково часто как среди девочек, так и среди мальчиков. Они имеют недоразвитую нижнюю челюсть, смещенный назад и западающий язык, изолированную расщелину неба. Гипоплазированная нижняя челюсть, дугообразное небо, особое расположение языка — основные признаки патологии, приводящие к обструкции респираторного тракта. Если размеры нижней челюсти соответствуют норме, то изменяется ее положение: она сильно смещается и делает большим угол основания черепа.

дети с синдромом Пьера Робена
У больных детей нарушаются жизненно важные функции организма — дыхание и глотание. Во время вдоха грудная клетка втягивается, малыши хрипят, а иногда вообще перестают дышать. В роддоме больным интубируют трахею для обеспечения дыхания. Синдром обструктивного апноэ – жизнеугрожающее состояние, нередко приводящее к внезапной смерти. Остановка дыхания обычно возникает в положении лежа на спине. При нарушении глотания питание осуществляться через желудочный зонд. Кормление обычным способом повышает риск попадания воды и пищи в дыхательные пути. Это может привести к развитию пневмонии или острого бронхита, которые угрожают жизни ребенка.

Недуг не имеет национальной и социальной принадлежности. Синдром встречается среди бедняков без соответствующего медицинского обслуживания, а также среди богатых и известных людей. Данная патология регистрируется в популяции довольно часто, но выявляемые признаки не рассматриваются в совокупности как проявления единого заболевания. Диагностируется аномалия Робена сразу после рождения ребенка. Начать лечение необходимо как можно раньше. После базовой терапии проводят пластику мягкого и твердого неба. Больные дети во время длительной реабилитации находятся под постоянным контролем медиков.
Этиопатогенез
Синдром Пьера Робена — генетически детерминированный недуг, передающийся детям от больных родителей по наследству. Это основная теория происхождения патологии. Существует менее распространенное мнение ученых-медиков, утверждающее, что синдром является изолированным. Эта форма развивается в результате сдавления нижней челюсти во время эмбриогенеза. Причинами компрессии являются: новообразования, кисты, очаги фиброза и рубцы на матке, многоплодная беременность, толчки или удары в матку во время вынашивания плода, вирусные и бактериальные инфекции в первом триместре беременности, нейротрофические расстройства, гиповитаминоз при беременности.

Нормальный процесс внутриутробного формирования челюстно-лицевой области при данной патологии нарушается. У больных неправильно формируется твердое небо, верхняя губа и ноздри. Аномальное соединение срединных выступов приводит к образованию вторичного неба, изменению формы лица, недоразвитию ротовых структур, неправильному расположению языка, несмыканию небесных отростков.
У больных мелкая ротовая полость не позволяет нормально разместиться языку, и он вынужден сместиться назад. Мягкое небо приобретает форму дуги. Язык располагается между глоточно-небными дужками и достигает синусов носа. Во время бодрствования малыша респираторный тракт остается свободным и проходимым для воздуха. Когда ребенок спит, приступы удушья возобновляются. При патологии нарушается не только дыхание, но процесс кормления. Без проведения терапевтических мероприятий это заболевание приводит к кахексии и смерти больных.
Кроме наследственной и изолированной теории формирования синдрома существует еще три физиологические гипотезы, объясняющие его происхождение:
- Механическая теория повествует о том, что недоразвитие нижней челюсти обусловлено высоким стоянием языка в полости рта, приводящим к формированию расщелин в небе и не полному закрытию небных пластин. Этот процесс происходит на 7-11-й неделе эмбриогенеза. У больных образуется классическая небная расщелина. Одной из причин патологии согласно механической теории является недостаток или отсутствие околоплодных вод.
- Неврологическое теория обусловлена нарушением проводимости нервных импульсов по волокнам подъязычного и глоточного нервных стволов, что было зафиксировано во время проведения электромиографического исследования.
- Наименее популярной и вероятной является теория, в основе которой лежит нарушение развития ромбэнцефалона во время эмбриогенеза.
Симптоматика
Основные клинические проявления синдрома:
- Неполноценное развитие нижней челюсти в медицине обозначается термином – нижняя микрогнатия. У больных нижняя зубная дуга втягивается на 10 мм за верхнюю арку. Этот симптом встречается практически у всех больных — в 90% случаев.
- Недостаточное развитие языка и его западание называется глоссоптозом. Симптом возникает в 75% случаев.
- Макроглоссия — увеличение размеров языка с ограничением его подвижности и функциональности. Встречается крайне редко.
- Анкилоглоссия — короткая уздечка языка и уплотнение тканей под ним вызывают трудности с его перемещением и кормлением. Этот относительно редкий признак отмечается в 15% случаев.
- Расщелина неба – «стрельчатое» небо.

К второстепенным и общим признакам патологии относятся:
- поверхностное, аритмичное и редкое дыхание,
- синюшность кожи,
- асфиксия при кормлении младенцев,
- затрудненное глотание,
- рвота,
- снижение или потеря слуха.
Больные дети беспокойны, они хрипят на вдохе, грудная клетка втягивается. Характерен внешний вид новорожденного – «птичье» лицо. Их рот до конца не закрывается, поэтому его называют «ртом акулы». Это связано с недоразвитием нижней челюсти различной степени выраженности: от незначительной до ее полного отсутствия.
Системные аномалии, характерные для данного синдрома:
- Аномалии глаз — миопия, гиперметропия, астигматизм, склероз роговицы, стеноз носослезного протока, врожденная катаракта.
- Нарушения строения слухового аппарата, пороки внутреннего уха, потеря слуха.
- Врожденные патологии сердца и сосудов — шумы в сердце, сужение легочного ствола, открытый артериальный проток, ДМПП, гипертензия в малом круге кровообращения.
- Пороки развития опорно-двигательного аппарата — сращение пальцев рук или ног, дисплазия фаланг, дополнительные пальцы на кистях или стопах, искривление пальцев, гипермобильность суставов, косолапость, искривление позвоночника, агенезия крестца и копчика, отсутствие некоторых конечностей.
- Патология ЦНС – эписиндром, водянка головного мозга, замедленное развитие речевой функции, умственная отсталость.
Недоразвитая ротовая полость и нижняя челюсть уродуют ребенка. Непроходимость респираторного тракта не дает малышу нормально дышать. Общее самочувствие улучшается во время плача и активного бодрствования, а затем опять наступает обструкция. Больные дети часто отказываются от еды и плачут, чтобы освободить дыхательные пути. Такие малыши истощенные, слабые и вялые. При отсутствии своевременного и правильного лечения прекратится функционирование внутренних органов и наступит смерть.
Стадии патологии:
- Легкая – дыхательная функция полностью сохраняется, процесс кормления не нарушается. Лечение консервативное в домашних условиях.
- Среднетяжелая – дыхание затрудняется, возникают проблемы с жеванием или глотанием, может наступить удушье. Лечение стационарное.
- Тяжелая – ребенка невозможно покормить, требуется желудочный зонд и наложение гастростомы. Он не может самостоятельно дышать. По показаниям проводится хирургическое вмешательство.
Видео: ребенок с синдромом Пьера Робена – при рождении и после лечения
Осложнения
Тяжелые осложнениями и негативными последствиями при синдроме Пьера Робена являются:
- Стридор — свистящее, шумное дыхание.
- Удушье.
- Нарушение речи.
- Частые отиты и лабиринтиты, ухудшение слуха.
- Неправильный прикус.
- Порок гортани.
- Прогрессирующие кардиологические заболевания.
- Снижение остроты зрения.
- Развитие заболеваний ЦНС.
- Смертельный исход.
Полноценное и своевременное лечение синдрома способно улучшить внешний вид и самочувствие ребенка.
Диагностика
Специалисты обычно безошибочно ставят диагноз больным, поскольку симптомы патологии достаточно явные и хорошо выраженные. Диагностика синдрома Пьера Робена основывается на характерных клинических проявлениях и не вызывает трудностей у врачей. Для исключения других врожденных патологий необходима консультация генетика.
Специалисты осматривают больных, изучают клинику и семейный анамнез. На основании полученных данных врачи ставят диагноз. В конце первого месяца жизни ребенка проводят нейросонографию — УЗИ мозга, позволяющее обнаружить аномалии в его строении.
Видео: синдром Пьера Робена на компьютерной томографии
Лечение
Дети с синдромом Пьера Робена прямо из роддома переводятся в реанимационное отделение, где проводится их длительное выхаживание.
Выявлением и лечением синдрома занимаются педиатры-неонатологи, ортодонты, генетики, хирурги. Сразу после рождения больного ребенка приступают к лечению. Легкая стадия заболевания требует выполнения основных рекомендаций по кормлению малыша: запрещено держать его горизонтально, чтобы не произошло удушье пищей. Правильному и быстрому росту нижней челюсти способствует вертикальное положение больного или его регулярное выкладывание на живот. Сила тяжести, воздействующая на нижнюю челюсть, обеспечивает ее рост и правильное формирование. Укладывание ребенка на живот способствует созданию необходимого давления в области головы, смещению языка на свое место и нормальному росту нижней челюсти.

Кормят больных детей с помощью ложечки, непрерывно контролируя дыхательные и глотательные движения. Перед этим очищают носовые проходы путем введения в ноздри ватных турунд, смоченных в вазелине. Детям с синдромом требуется увеличить суточную калорийность питания. Для этого смешивают грудное молоко и искусственную смесь. Кормить таких детей следует с перерывами, которые позволят отдышаться и очистить дыхательные пути. Поглаживания подбородка малыша помогают ему поглощать молоко.
Медикаментозная терапия заключается в назначении следующих групп препаратов:
- Барбитураты – успокоительные средства, обладающие противосудорожным и снотворным действием: «Фенобарбитал», «Гексобарбитал», «Бутизол».
- Производные бензодиазепина – мощные миорелаксанты и анксиолитики, оказывающие противосудорожное действие: «Клоназепам», «Нитразепам», «Ривотрил».
- Транквилизаторы – успокоительные препараты, снимающие судороги, тревогу, страх и облегчающие общее состояние пациента: «Сибазон», «Реланиум», «Седуксен».
- Ноотропные препараты, улучшающие мозговое кровообращение и стимулирующие высшие функции ЦНС – «Кортексин», «Пирацетам», «Винпоцетин».
- Сердечные и дыхательные аналептики стимулируют дыхательный центр и нормализуют сосудистый тонус – «Кордиамин», «Камфора», «Кофеин».
- Кислородотерапия.
При выраженной гипоплазии нижней челюсти требуется проведение операции, в процессе которой выводят запавший язык и фиксируют его в правильном положении.
Виды операций:
- Глоссопексия выполняется для предотвращения западения языка. Этот метод лечения считается высокоэффективным и полностью безвредным. Выполняют вмешательство двумя способами: путем протягивания серебряной нити через нижнюю часть десны и нижнюю губу, или же протягивая ее через основание языка и обе щеки. Кормление ребенка осуществляется через гастростому.
- Дистракционный остеосинтез — создание на нижней челюсти двух отверстий, в которые устанавливают штифты. Их фиксируют на голове с помощью особого механизма. Постепенно аппаратами разводят закрепленные части на нужную величину. Благодаря усилению, с которым воздействует механизм на фрагменты челюсти, кость удлиняется. Этот метод также дает хороший эффект. Лечение проводят месяц, а затем делают перерыв для полного заживления кости. Штифты фиксируют в новом месте и повторяют курс лечения. Снимают конструкцию примерно через 3 месяца, когда костная мозоль превратится в полноценную кость.
- Операцию по устранению расщелины на небе проводят в два этапа. Пластика мягкого и твердого неба показана в разном возрасте. В первом случае операцию выполняют детям до года, во втором — в возрасте 5—6 лет. Хирурги накрывают расщелину неба на всем протяжении, удлиняют мягкое небо и сужают средний отдел глотки.
- В запущенных случаях интубируют трахею, благодаря чему больные начинают дышать. В данной операции нуждается всего 1% детей с заболеванием тяжелой степени. Трахеостому закрывают примерно в 3 года. Длительное зондовое питание и трахеостомия чреваты развитием пневмонии и ослаблением глотательного рефлекса.

результаты оперативного лечения синдрома Пьера Робена
В течение всего лечебного процесса больной ребенок находится на контроле у специалистов в области офтальмологии, логопедии, оториноларингологии, стоматологии. Своевременное обращение за медицинской помощью повышает шансы на полное устранение симптомов патологии. Перечисленные методы консервативного и оперативного лечения позволяют постепенно нормализовать размеры и форму нижней челюсти у большинства больных детей.
Профилактика и прогноз
Чтобы предупредить развитие синдрома Пьера Робена, необходимо устранить все возможные негативные факторы, оказывающее воздействие на внутриутробное развитие плода — удаление новообразований в матке, коррекция гормонального фона. Пренатальная диагностика также позволяет избежать появления на свет больных детей. Для этого проводят УЗИ беременной женщины, скрининг сывороточных маркеров, биопсию ворсин хориона, исследование околоплодных вод и пупочной крови. Не менее важную роль играет прохождение регулярных осмотров у гинеколога, своевременная постановка на учет беременной женщины.
Прогноз патологии у новорожденных детей в целом неблагоприятный. Смерть часто наступает от асфиксии или многочисленных инфекций. У годовалых детей в результате лечения дыхание нормализуется, челюсть становится на место и принимает нужные размеры. У пациентов в возрасте 1-2 лет прогноз относительно благоприятный.
Источник
Медицинский эксперт статьи
х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Пьера Робена, также известный в медицине под названием аномалия Робена, является врожденной патологией развития челюстной части лица. Свое название заболевание получило в честь французского стоматолога П. Робена, который впервые описал все его признаки. Lannelongue и Menard впервые описали синдром Пьера Робина в 1891 г. в своем докладе на примере 2-х пациентов с микрогнатией, расщелиной нёба и ретроглоссоптозом. В 1926 году Пьер-Робин опубликовал случай заболеввания у младенца с признаками классического синдромом. До 1974 года триада признаков была известна как синдром Робена-Пьера. Тем не менее, этот синдром сейчас используется для описания пороков формообразования при одновременном наличии множественных аномалий.
Код по МКБ-10
Q87.0 Синдромы врожденных аномалий, влияющие преимущественно на внешний вид лица
Эпидемиология
Это гетерогенный врожденный дефект, который имеет распространенность 1 на 8500 живорожденных. Соотношение мужчин к женщинам составляет 1:1, за исключением Х-хромосомой формы.
Среди этих больных, у 50% младенцев расщелина на мягком небе является неполной, остальные рождаются с дугообразным и необычайно высоким небом, но без расщелины.
[1], [2], [3], [4], [5], [6], [7], [8], [9]
Причины Синдрома Пьера Робена
Рассматривается возможность аутосомно-рецессивного наследования болезни. Различают два вида синдрома в зависимости от этиологии: изолированный и генетически детерминированный. Изолированный вид развивается из-за компрессии нижней части челюсти во период эмбрионального развития. Компрессия может развиваться вследствие:
- Наличие в матке локальных уплотнений (кисты, рубцы, опухоли).
- Многоплодная беременность.
Также развитие челюсти у плода может нарушаться при:
- Вирусных инфекциях, которые перенесла будущая мать во время беременности.
- Нейротрофических нарушениях.
- Недостаточном количестве фолиевой кислоты в организме беременной женщины.
[10], [11], [12], [13], [14]
Патогенез
Синдром Пьера Робена проявляется из-за эмбриональных нарушений, которые вызываются самыми разнообразными патологиями в дородовом периоде.
Существует три патофизиологические теории, которые могут объяснить возникновение синдрома Пьера Робена.
Механическая теория: Эта теория наиболее вероятная. Недоразвитие нижнечелюстного аппарата происходит между 7-й и 11-й неделями беременности. Высокое стояние языка в ротовой полости приводит к образования расщелин в небе, из-за этого не происходит закрытие небных пластинок. Эта теория объясняет классическую перевернутую U-образную расселину и отсутствие связанной с ней заячьей губы. В этиологии определенную роль может играть олигогидрамнион, так как отсутствие амниотической жидкости может привести к деформации подбородка и последующего сдавление языка между небных пластинок.
Неврологическое теория: Задержка в неврологическом развитии была отмечена при проведении электромиографии мышц язычка и глоточных столбов, и вкуса из-за задержки проводимости в подъязычном нерве.
Теория диснейрорегуляции ромбовидного мозга: Эта теория основана на нарушении развития ромбовидного мозга в процессе онтогенеза.
Недостаточное развитие нижней части челюсти ребенка приводит к тому, что ротовая полость значительно уменьшается. Это, в свою очередь, вызывает так называемую псевдомакроглоссию, то есть язык смещается к задней части стенки глотки. Такая патология приводит к развитию обструкции дыхательного пути.
Пока младенец плачет или двигается, проходимость дыхательного пути остается нормальной, но как только он засыпает, снова возникает обструкция.
Из-за респираторных нарушений процесс кормления младенца сильно затруднен. В это время практически всегда возникает обструкция дыхательного пути. Если не применять лечебную коррекцию, то такая патология может привести к сильному истощению всего организма и даже к летальному исходу.
[15], [16], [17], [18], [19], [20]
Симптомы Синдрома Пьера Робена
Заболевание отличается трема основными признаками:
- Нижняя микрогнатия (недостаточное развитие нижней части челюсти, встречается в 91,7% случаев заболевания). Она характеризуется втягиванием нижней зубной дуги на 10-12 мм позади верхней арки. Нижняя челюсть имеет небольшое тело, тупой угол. Ребенок достигает нормального развития приблизительно в возрасте 5-6 лет.
- Глоссоптоз (западание языка по причине его недостаточного развития, отмечается в 70-85% случаев).
- Макроглоссия и анкилоглоссия относительно редкие признаки, отмечаются в 10-15% случаев.
- На небе появляется расщелина.
- Брадипноэ и диспноэ.
- Легкий цианоз.
- Асфиксия, которая чаще всего проявляется во время попыток покормить младенца.
- Глотание невозможно или сильно затруднено.
- Позывы к рвоте.
- Аурикулярные аномалии в 75% случаев.
- Потеря слуха проводящего характера встречается у 60% больных, в то время как атрезия наружного слухового канала встречается только у 5% пациентов, недостаточная пневматизация сосцевидного полости височной кости.
- Аномалии внутреннего уха (аплазия боковых полукруглых каналов, большого вестибулярного акведука, потеря волосковых клеток улитки).
- Носовые пороки развития являются нечастыми и представлены в основном из аномалий корня носа.
- Стоматологические пороки развития встречаются в 30% случаев. Ларингомаляция и небно-глоточная недостаточность наблюдаются приблизительно у 10-15% пациентов с синдромом Пьера Робина.
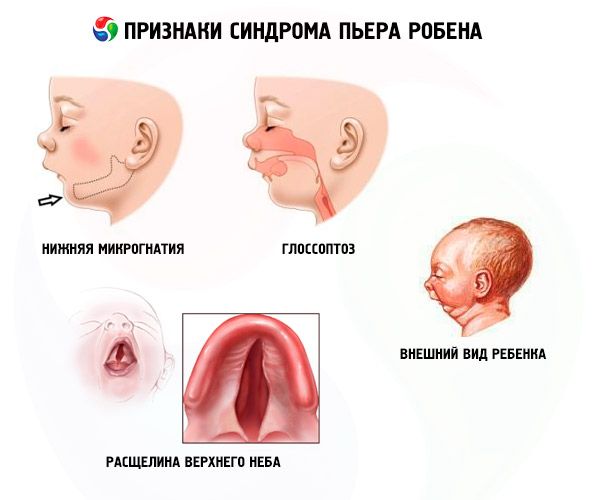
Системные признаки синдрома Пьера Робина
Системные аномалии развития описаны в 10-85% зарегистрированных случаев.
Глазные аномалии встречаются в 10-30% пациентов. Могут встречаться: дальнозоркость, близорукость, астигматизм, склероз роговицы и стеноз носослезного протока.
Сердечно-сосудистые патологии: доброкачественные сердечные шумы, стеноз легочной артерии, открытый артериальный проток, овальное окно, дефект межпредсердной перегородки и легочная гипертензия. Их распространенность варьирует в от 5-58%.
Аномалии, связанные с опорно-двигательным аппаратом (70-80% случаев): синдактилия, диспластические фаланги, полидактилия, клинодактилия, гиперподвижность суставов и олигодактилия верхних конечностей. Аномалии развития нижних конечностей: аномалии стоп (косолапость, аддукция плюсны), бедренные пороки развития (вальгусный или варусный таз, короткие бедра), аномалии бедра (врожденный вывих, контрактуры), аномалии коленного сустава (GENU VALGUS, синхондроз). Пороки развития позвоночного столба: сколиоз, кифоз, лордоз, позвоночная дисплазии, агенезия крестца и копчиковой пазухи.
Патология центральной нервной системы: эпилепсия, задержки развития нервной системы, гидроцефалия. Частота дефектов ЦНС составляет около 50%.
Мочеполовые аномалии: не опустившиеся семенники (25%), гидронефроз (15%), а также водянка яичка (10%).
Ассоциированные синдромы и состояния: синдром Стиклера, синдром трисомии 11q, трисомии 18, синдром удаления 4q, ревматоидная артропатия, гипохондроплазия, синдром Мебиуса.
Стадии
Существует три стадии тяжести заболевания, которые зависят от состояния дыхательных путей ребенка:
- Легкая – есть небольшие проблемы с кормлением, но дыхание почти не затруднено. Лечение проводят в амбулаторно.
- Средняя – дыхание умеренно затруднено, кормление ребенка умеренно трудное. Лечение проводят в стационаре.
- Тяжелая – дыхание очень затруднено, ребенка невозможно нормально кормить. Необходимо использовать специальные приспособления (интраназальный зонд).
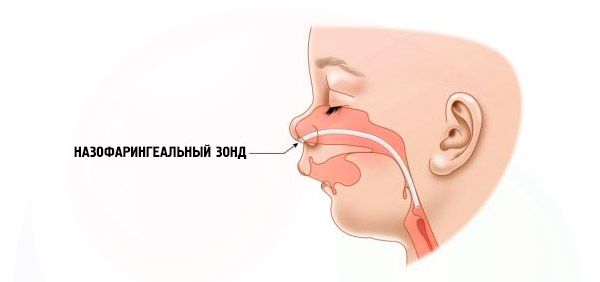
[21], [22], [23], [24]
Осложнения и последствия
Сочетание микрогнатии и глоссоптоза может привести к тяжелым респираторным осложнениям и проблемам во время кормления ребенка.
Синдром Пьера Робена вызывает следующие осложнения:
- Стридозное дыхание из-за обструкции дыхательного пути. Ларингомаляция или даже асфиксия во сне.
- Психомоторное развитие ребенка сильно отстает от сверстников.
- Физическое развитие также отстает.
- Речь у больных нарушена.
- Частые инфекционные заболевания уха, которые становятся хроническими и приводят к нарушениям слуха.
- Синдром обструктивного апноэ, наступление смерти во сне варьирует в 14-91% случаев.
- Проблемы с зубами.
[25], [26], [27], [28], [29], [30]
Диагностика Синдрома Пьера Робена
Диагностика синдрома Пьера Робена трудностей не вызывает. Она основывается на клинических проявлениях. Чтобы исключить другие патологии, очень важно проконсультироваться у генетика.
У детей с врожденной аномалией Робена с самого рождения нарушено дыхание из-за постоянного западания языка. Младенец беспокойно себя ведет, кожные покровы его синюшные, при вдохах из грудной клетки вырывается хрип. В процессе кормления могут наступать удушья. Диагноз можно поставить также по необычному внешнему виду ребенка – «птичьему лицу». Часто у больных развиваются и другие пороки: миопия, катаракта, патология мочеполовой системы, патология сердца, аномалии развития позвоночника.
По этим клиническим проявлениям поставить правильный диагноз специалисту не составит труда.
[31], [32], [33], [34], [35], [36]
Лечение Синдрома Пьера Робена
Лечение проводят сразу же после рождения ребенка с синдромом Пьера Робена. Если заболевание носит легкий характер, то для улучшения состояния больного необходимо постоянно держать ребенка вертикально или лежа на животике. Голову младенца нужно наклонять к груди. В процессе кормления не рекомендовано держать ребенка в горизонтальном положении, чтобы пища не попадала в дыхательные пути.
Если недостаточное развитие нижней части челюсти выражено довольно сильно, применяется оперативное вмешательство для выведения западающего языка в нормальное физиологическое положение. При тяжелых случаях язык подтягивают и фиксируют на нижней губе. При очень тяжелых случаях необходимо проведения трахеостомии, глоссопексии, дистракционного остеогенеза нижней челюсти.
Также применяется и консервативное лечение.
Лекарства
Фенобарбитал. Снотворный и седативный препарат, отличается противосудорожным эффектом. В каждой таблетке находится 100 мл фенобарбитала. Дозировка является индивидуальной, так как зависит от степени тяжести болезни и состояния ребенка. Пациентам с печеночной недостаточностью, гиперкинезом, анемией, миастенией, порфирией, сахарным диабетом, депрессией, непереносимостью компонентов препарат запрещен. При приеме возможны следующие симптомы: головокружение, астения, галлюцинации, агранулоцитоз, тошнота, пониженное артериальное давление, аллергия.
Клоназепам. Препарат, который назначается для лечения эпилепсии. В лекарстве находится активное вещество клоназепам, которое является производным бензодиазепина. Отличается противосудорожным, анксиолитическим и миорелаксирующим эффектом. Доза устанавливается лечащим врачом, но не должна превышать максимальной – 250 мкг в день. Не принимать при бессоннице, мышечном гипертонусе, психомоторном возбуждении, панических расстройствах. При приеме возможны следующие симптомы: заторможенность, тошнота, дисменорея, головная боль, лейкопения, задержка или недержание мочи, алопеция, аллергия.
Сибазон. Выпускается в форме раствора и ректальных таблеток. Активным веществом является производным бензодиазепина (сибазон). Отличается седативным, анксиолитическим, противосудорожным эффектом. Дозировка является индивидуальной. Пациентам с хронической гиперкапнией, миастенией, непереносимостью бензодиазепинов принимать препарат запрещено. При употреблении средства возможно развитие таких симптомов: тошнота, запоры, головная боль, головокружение, икота, недержание мочеиспускания, аллергия.
Кортексин лиофилизат. Препарат с ноотропным действием. В лекарстве находится комплекс полипептидных фракций растворимых в воде и глицин. Дозировка является индивидуальной и назначается лечащим врачом в соответствии с состоянием больного. Пациентам с непереносимостью кортексина принимать препарат запрещено. Средство может вызывать аллергические реакции.
Физиотерапевтическое лечение
Как правило, при легких стадиях синдрома проводится позиционная терапия, когда ребенка укладывают на живот в вертикальном положении до тех пор, пока сила тяжести не заставит нижнюю часть челюсти расти правильно.
Оперативное лечение
Оперативное лечение используется, в первую очередь, для коррекции глоссоптоза. Существует несколько методов:
- Поддерживание с помощью серебряной нити языка. Нить проводится через нижнюю часть десны и нижнюю губу. Метод носит название Дугласа.
- Метод Духамеля – толстую серебряную нить проводят через основание языка пациента и две щеки. Использовать не дольше тридцати дней.
- Ортопедические аппараты для вытягивания и фиксации языка.
- В возрасте одного года можно проводить операцию по устранению расщелины на небе.
Профилактика
Единственным методом профилактики синдрома Пьера Робена является устранение возможных негативных факторов в дородовом периоде развития плода, пренатальная диагностика.
[37], [38], [39], [40], [41], [42], [43]
Прогноз
Прогноз и течение заболевания – тяжелые. Чаще всего в первые же дни жизни при средней и тяжелой стадии заболевания наступает смерть (причина – асфиксия). Также риск летального исхода в первый год достаточно высокий из-за многочисленных инфекций.
У пациентов в возрасте после двух лет – прогноз благоприятный.
[44], [45]
Источник