
Синдром тернера признаки на узи

Синдром Тернера у плода. Диагностика и прогноз синдрома Тернера
Хромосомное заболевание, выявляемое у женщин, было описано Н.Н. Turner в 1938 году и характеризовалось наличием одной Х-хромосомы (45,Х0).
Синонимы. Моносомия X и синдром Ульриха-Тернера (Ullrich-Turner).
Распространенность. От 2,5 до 5,5 на 10 000 новорожденных девочек у белокожих людей. Среди жителей Японии этот показатель составляет от 7 до 21 на 10 000 новорожденных девочек. Однако распространенность по отношению к общему числу беременностей считается еще более высокой, поскольку синдром Тернера (Turner) обнаруживается в 25% случаев спонтанных абортов, вызванных хромосомными аномалиями.
Этиология. Отсутствие одной половой хромосомы, в большинстве случаев передающейся от отца, происходит чаще всего спорадически, и от 8 до 16% всех заболеваний обусловлены мозаицизмом. Не обнаружено связи возраста матери с возникновением этого вида анеуплоидии.
Риск рецидива. Учитывая, что синдром Тернера (Turner) в большинстве случаев возникает спорадически, шансы рецидива исключительно низки, хотя точно неизвестны.
Диагностика. Кистозная гигрома является наиболее характерным признаком данного синдрома в пренатальном периоде. В большинстве наблюдений она бывает представлена большим образованием кистозного строения с перегородками, которое располагается в области задней поверхности шеи и имеет общую лимфатическую циркуляцию. Может иметь место спонтанное исчезновение гигромы, что во многих случаях приводит к формированию кожных шейных сладок.
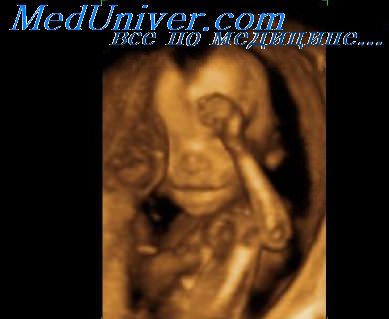
Другие распространенные признаки включают лимфатический отек, особенно тыльных поверхностей кистей и стоп или в более тяжелых случаях – анасарку, водянку, а также короткий шейный отдел позвоночника, задержку внутриутробного развития, торчащие ушные раковины, подковообразную почку, пороки сердца (коарктация аорты и двухстворчатый аотральный клапан), дисплазию костей и низкий рост.
Дисгенезия яичников имеет место в 90% случаев, что обусловливает отсутствие менструаций и бесплодие во взрослом возрасте. Также часто встречаются различные степени задержки умственного и физического развития. Имеются сообщения о повышенном уровне субъединицы хорионического гонадотропина в сыворотке крови у матери при наличии синдрома Тернера (Turner) у плода.
Дифференциальный диагноз. Необходимо исключить кистозные образования в области головы и шеи, такие как цефалоцеле и менингоцеле. Обычно эти состояния сочетаются с гидроцефалией и дефектами развития черепа и позвоночника. При дифференциальной диагностике следует использовать оценку характера угла, возникающего на границе соединения опухоли с окружающей кожей.
Кистозная гигрома приподнимает прилежащие ткани и имеет тенденцию формировать пологий угол в этой области. В то время как цефалоцеле, которое является грыжей и выпячивается через кожный дефект, характеризуется острым углом в области «примыкания» к поверхности кожи. Кистозные гигромы встречаются при синдроме Роберта (Robert) и других аутосом-но-рецессивных заболеваниях. Хромосомный анализ позволяет исключить эти состояния.
Генетические нарушения. За возникновение данного состояния также могут быть ответственны отсутствие или структурные аномалии одной из Х-хромосом, такие как делеция региона, детерминирующего пол (XY Yp делеция), или состояния, связанные с мозаицизмом (45,Х/46,ХХ или 45X/46XY).
Прогноз. Во многих случаях происходит антенатальная гибель, часто обусловленная водянкой, которая является основным осложнением в этом периоде. Для выживших прогноз будет зависеть от тяжести имеющихся сочетанных аномалий. Мозаицизм характеризуется тенденцией к более благоприятному прогнозу, и некоторые случаи с неполными клиническими проявлениями синдрома остаются недиагностированными в течение многих лет.
– Также рекомендуем “Трансфузионный фето-фетальный синдром. Диагностика и механизмы развития фето-фетального синдрома”
Оглавление темы “Врожденные синдромы плода”:
1. Синдром Робена. Септо-оптическая дисплазия у плода
2. Синдромы коротких ребер – полидактилии. Диагностика и прогноз синдрома коротких ребер
3. Сиреномелия. Синдром русалки у плода
4. Танатофорная дисплазия. Диагностика и прогноз танатоформной дисплазии
5. Синдромы тромбоцитопении. Синдром аплазии лучевой кости
6. Туберозный склероз у плода. Склероз Бурневилля
7. Синдром Тернера у плода. Диагностика и прогноз синдрома Тернера
8. Трансфузионный фето-фетальный синдром. Диагностика и механизмы развития фето-фетального синдрома
9. Дифференциация фето-фетального синдрома. Прогноз и тактика при фето-фетальном синдроме
10. Сочетание аномалий VACTERL. Секвенция VATER
Источник
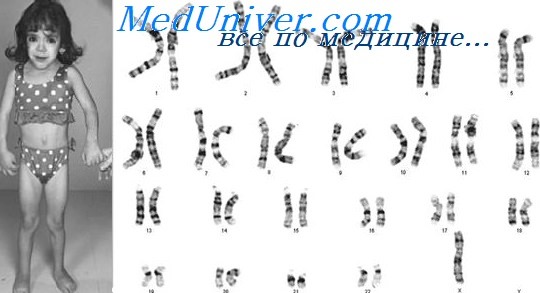
Синдром Тернера: причины, диагностика, лечениеЭтиология и встречаемость синдрома Тернера. Синдром Тернера — панэтническое заболевание, вызываемое полным или частичным отсутствием второй Х-хромосомы у женщин. Его встречаемость составляет от 1 на 2000 до 1 на 5000 среди живорожденных девочек. Около 50% случаев синдрома Тернера вызваны кариотипом 45,Х, 25% — структурной аномалией второй Х-хромосомы, и 25% — мозаицизмом 45,Х. Моногамия по Х-хромосоме может возникать при нарушении перехода половой хромосомы в одну из гамет или при потере половой хромосомы в зиготе или раннем эмбрионе. Наиболее частая причина кариотипа 45,Х — нарушение передачи отцовской половой хромосомы в гамету; 70-80% пациенток с кариотипом 45,Х оплодотворены сперматозоидом с недостающей половой хромосомой. Вероятная причина мозаицизма 45,Х — утрата половой хромосомы в клетке раннего эмбриона. Патогенез синдрома ТернераМеханизм формирования клинической картины синдрома Тернера у девочек с моносомей по Х-хромосоме мало понятен. Х-хромосома содержит множество локусов, не подвергающихся инактивации, некоторые из них необходимы для функционирования яичников и женской фертильности. Хотя для развития овоцита достаточно одной Х-хромосомы, для их функционирования необходимы две Х-хромосомы. Следовательно, при отсутствии второй Х-хромосомы, овоциты у плодов и новорожденных с синдромом Тернера деградируют, а их яичники атрофируются в полоски соединительной ткани. Генетическая основа других признаков синдрома Тернера, например, кистозной гигромы, лимфоотека, широкой грудной клетки, пороков сердца, аномалий почек и нейросенсорной тугоухости, не определена, но, возможно, отражает гаплонедостаточность одного или более Х-сцепленных генов, в норме не подвергающихся инактивации у женщин. Фенотип и развитие синдрома ТернераХотя плоды с кариотипом 45,Х встречаются при 1-2% всех беременностей, заканчиваются живорожденным младенцем с кариотипом 45,Х менее 1% родов. Учитывая мягкий фенотип, наблюдаемый у пациенток с синдромом Тернера, обращает на себя внимание столь высокий показатель выкидышей, указывая на важность второй половой хромосомы для внутриутробного выживания. Все пациенты с синдромом Тернера имеют низкий рост, и более 90% —дисгенезию яичников. Дисгенезия яичников достаточно выраженная, поэтому только у 10-20% пациентов пубертат развивается самостоятельно (молочные железы и лобковое оволосение) и только 2-5% имеют самостоятельные менструации. Большинство больных также имеет физические аномалии, например, складчатую шею, низкий рост волос на затылке, широкую грудную клетку, пороки сердца и почек, нейросенсорную тугоухость, отеки кистей и стоп и диспластичные ногти. Почти у 50% пациенток отмечают двустворчатый аортальный клапан, и, следовательно, повышенный риск расширения и расслаивающей аневризмы корня аорты; почти у 60% имеются аномалии почек и повышенный риск дисфункции почек.
У большинства пациенток интеллектуальное развитие нормальное. Если есть интеллектуальная задержка, обычно обнаруживается структурная аномалия Х-хромосомы. Больные с синдромом Тернера обычно стеснительны и замкнуты (см. главу 6). Помимо осложнений, вызванных врожденными аномалиями, у женщин с синдромом Тернера чаще встречаются остеопоротические переломы костей, тиреоидит, сахарный диабет I и II типа, колиты и заболевания сердечно-сосудистой системы. Причины сахарного диабета, нарушений щитовидной железы и колитов неясны. Вероятно, в основном за остеопороз и повышенную встречаемость атеросклероза, ишемических заболеваний сердца и инсульты ответственна эстрогенная недостаточность, хотя возможно, что сахарный диабет усиливает влияние недостатка эстрогенов на сердечно-сосудистую систему. Особенности фенотипических проявлений синдрома Тернера: Лечение синдрома ТернераЕсли рост пациентки с синдромом Тернера отстает ниже 5-го процентиля, обычно назначают соматотропный гормон, применяемый до достижения костного возраста, соответствующего 15 годам. В среднем это лечение дает прибавку роста в 10 см; прибавка роста тем меньше, чем позже начато лечение гормоном роста. Параллельная терапия эстрогенами уменьшает эффективность гормона роста. Терапию эстрогенами обычно начинают в возрасте 14-15 лет, для обеспечения развития вторичных половых признаков и снижения риска остеопороза. Прогестерон для вызывания менструаций добавляют к лечению либо во время первого вагинального кровотечения, либо на втором году терапии эстрогенами. Кроме того, медицинская помощь обычно включает следующие виды диагностики: эхокардиографию с целью оценки расширения корня аорты и клапанных заболеваний сердца, УЗИ почек для исключения врожденных аномалий и нагрузочный тест с глюкозой для выявления сахарного диабета. У пациенток с полной дисгенезией яичников не бывает спонтанных овуляций и беременности. Однако если позволяет состояние сердечно-сосудистой системы и почек, женщины с синдромом Тернера могут иметь детей в случае подсадки оплодотворенной яйцеклетки. Риски наследования синдрома ТернераСиндром Тернера не связан с возрастом матери или отца. Хотя описано несколько случаев повторения в семье, обычно синдром Тернера спорадический, и эмпирический риск повторения при будущих беременностях не превышает общепопуляционного. Если синдром Тернера заподозрен на основе УЗИ плода, например, выявлена кистозная гигрома, диагноз следует подтвердить кариотипированием ворсин хориона или амниоцитов. Среди спонтанно менструирующих женщин с синдром Тернера описано несколько случаев наступления беременности. У одного из трех родившихся детей отмечены врожденные аномалии, такие как врожденные пороки сердца, синдром Дауна и spina bifida. Очевидно, повышенный риск врожденных аномалий может быть следствием выборочного смещения в публикациях, так как беременность редко встречается при синдроме Тернера. Если повышенный риск аномалий — реальный факт, то его причина неизвестна. Пример синдрома Тернера. Л.В., девушка 14 лет, направлена в клинику эндокринологии для обследования по поводу отсутствия вторичных половых признаков (менструаций и развития молочных желез). Хотя Л.В. родилась с небольшой массой тела, она была полностью здорова и имела нормальный интеллект. Другие члены семьи аналогичных проблем не имели. Данные обследования оказались нормальными, за исключением низкого роста, I стадии полового развития по Таннеру и широкой грудной клетки с широко расположенными сосками. После краткого обсуждения возможных причин задержки роста и полового развития врач назначил определение уровня фолликулостимулирующего гормона, гормона роста (соматотропина), анализ костного возраста и анализ хромосом. Эти тесты показали нормальный уровень гормона роста, повышение уровня фолликулостимулирующего гормона и аномальный кариотип (45,Х). Врач дал заключение, что у девочки синдром Тернера. Ей был назначен гормон роста; годом позже она начала терапию эстрогенами и прогестероном для стимуляции развития вторичных половых признаков. – Также рекомендуем “Пигментная ксеродерма: причины, диагностика, лечение” Оглавление темы “Генетика”:
|
Источник
Медицинский эксперт статьи
х
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Тернера (синдром Шерешевского-Тёрнера, синдром Бонневи-Ульриха, Синдром 45, Х0) является следствием полного или частичного отсутствия одной из двух половых хромосом, фенотипически определяется женский пол. Диагноз основывается на клинических проявлениях и подтверждается исследованием кариотипа. Лечение зависит от проявлений и может включать оперативное лечение при пороках сердца, и часто терапию гормоном роста при низкорослости и заместительную терапию эстрогеном при отсутствии пубертата.
Клинически впервые описан отечественным эндокринологом Н. А. Шерешевским в 1926 г. Цитогенетически синдром верифицирован в 1959 г. Популяционная частота 1 : 5000 лиц женского пола.
[1], [2], [3], [4], [5], [6]
Код по МКБ-10
Q96 Синдром Тернера
Q96.8 Другие варианты синдрома Тернера
Q96.9 Синдром Тернера неуточненный
Эпидемиология синдрома Шерешевского-Тёрнера
Синдром Шерешевского-Тёрнера встречается с частотой примерно 1 /4000 живорождений и является наиболее распространенной аномалией половых хромосом у женщин. В то же время 99 % беременностей с кариотипом плода 45, X заканчиваются спонтанным абортом.
[7], [8], [9], [10], [11]
Что вызывает синдром Шерешевского-Тёрнера?
Примерно у 50 % пациенток с синдромом Шерешевского-Тёрнера отмечается кариотип 45, X; примерно в 80 % случаев теряется отцовская Х-хромосома. Большая часть оставшихся 50 % мозаична (например, 45, Х/46, XX или 45, Х/47, XXX). Среди мозаичных пациентов фенотип может варьировать от типичного для синдрома Тернера до нормального. Иногда у девочек с синдромом Шерешевского-Тёрнера могут отмечаться одна нормальная Х-хромосома и одна Х-кольцевая хромосома; для того чтобы произошло формирование кольцевой хромосомы, она должна потерять по участку с короткого и длинного плеча. Некоторые девочки с синдромом Тернера могут иметь одну нормальную Ххромосому и одну изохромосому, состоящую из двух длинных плеч Х-хромосомы, которая сформировалась после потери короткого плеча. У таких девочек отмечается склонность иметь многие из фенотипических черт синдрома Тернера; таким образом кажется, что деления короткого плеча Х-хромосомы играет важную роль в формировании фенотипа синдрома Шерешевского-Тёрнера.
Патогенез синдрома Шерешевского-Тёрнера
Отсутствие или изменение половой хромосомы приводит к нарушению созревания яичников, отсутствию или позднему частичному половому созреванию и бесплодию.
Симптомы синдрома Шерешевского-Тёрнера
У многих новорожденных отмечаются только очень легкие проявления; однако у некоторых наблюдаются выраженная дорсальная лимфедема кистей рук и ступней, а также лимфедема или кожные складки на задней поверхности шеи. Другие распространенные аномалии включают крыловидные складки шеи, широкую грудную клетку и втянутые соски. У пораженных девочек отмечается низкий рост по сравнению с членами семьи. Менее частыми признаками являются низкая линия роста волос на задней поверхности шеи, птоз, множественные пигментные невусы, короткие четвертые пястная и плюсневая кости, выступающие подушечки пальцев с завитками на концах пальцев, а также гипоплазия ногтей. Также отмечаются cubitus valgus (вальгусное отклонение в локтевом суставе).
Распространенные аномалии сердца включают коарктацию аорты и двухстворчатый клапан аорты. Часто с возрастом развивается гипертензия, даже в отсутствие коарктации. Нередко встречаются аномалии почек и гемангиомы. Иногда в ЖКТ обнаруживают телеангиэктазию, с развивающимися желудочнокишечными кровотечениями или потерей белка.
Дисгенезия гонад (яичники замещаются двухсторонними тяжами фиброзной стромы с отсутствием развивающихся половых клеток) отмечается у 90 % больных, приводя к отсутствию пубертата, отсутствию увеличения грудных желез, аменорее. В то же время у 5-10 % пораженных девочек спонтанно происходит менархе, и очень редко пораженные женщины фертильны и имеют детей.
Задержка умственного развития отмечается редко, но у многих пациентов наблюдается снижение некоторых перцептивных возможностей и вследствие этого низкие оценки в невербальных тестах и по математике, даже несмотря на то, что баллы, полученные за вербальный компонент тестов на интеллект, средние или даже высокие.
- Задержка роста, часто с рождения (100%).
- Гонадальный дисгенез с аменореей и стерильностью.
- Лимфатический отёк тыла кистей и стоп (40%).
- Широкая грудная клетка с комбинированной деформацией грудины.
- Широко расставленные, гипоплазированные и инвертированные соски (80%).
- Аномальные по форме и оттопыренные ушные раковины (80%).
- Низкий уровень роста волос.
- Короткая шея с избытком кожи и крыловидными складками (80%).
- Cubitus valgus(70%).
- Узкие, гипервогнутые и вдавленные ногти (70%).
- Врождённые пороки почек (60%).
- Тугоухость (50%).
- Врождённые пороки сердца и аорты (коарктация аорты и патология клапанов, расширение и расслоение аорты) (20-40%).
- Идиопатическая артериальная гипертензия (АГ) (27%).
Диагностика синдрома Шерешевского-Тёрнера
У новорожденных диагноз можно заподозрить при наличии лимфедемы или крыловидной складки шеи. В отсутствие этих изменений диагноз у некоторых детей выявляют позже на основании низкого роста, аменореи и отсутствия пубертата. Диагноз подтверждается исследованием кариотипа. Для обнаружения врожденных пороков сердца показано проведение эхокардиографии или МРТ.
Цитогенетический анализ и исследования с Y-специфичным зондом проводят всем лицам с дисгенезией гонад для исключения мозаицизма с наличием клеточной линии с кариотипом 46, XY (45, X/46XY). У таких пациентов обычно женский фенотип с различными чертами синдрома Тернера. Они находятся в группе повышенного риска по развитию злокачественных новообразований гонад, особенно гонадобластомы, поэтому для профилактики сразу после определения диагноза следует удалить гонады.
[12], [13], [14], [15], [16], [17]
Физическое обследование
Диагноз ставят на основании характерной клинической картины: короткая шея с избытком кожи и крыловидными складками у новорождённых девочек, лимфатический отёк кистей и/или стоп, врождённые пороки левого сердца или аорты (особенно коарктация аорты), задержка роста и полового развития в пубертатный период у девочек.
Лабораторные исследования
Цитогенетическое исследование верифицирует отсутствие Х-хромосомы или её структурные изменения.
Какие анализы необходимы?
Лечение синдрома Шерешевского-Тёрнера
Не существует специфического лечения генетического нарушения. Коарктацию аорты обычно лечат оперативно. При других пороках сердца показаны динамическое наблюдение и хирургическая коррекция при необходимости. При лимфедеме применяют компрессионные чулки.
Терапию рекомбинантным человеческим гормоном роста могут начинать, если рост менее 5 перцентилей, начальная доза составляет 0,05 мг/кг подкожно один раз в день. Как правило, в возрасте 12-13 лет для инициации пубертата необходима заместительная терапия эстрогеном в виде конъюгированных эстрогенов по 0,3 мг внутрь или микронизированного эстрадиола по 0,5 мг один раз в день. После этого назначают оральные контрацептивы, содержащие прогестин, для поддержания вторичных половых признаков. Гормон роста продолжают вводить вместе с терапией половыми гормонами до закрытия зон роста, когда терапию гормоном роста прекращают. Продолжение заместительной терапии половыми гормонами помогает достичь оптимальную плотность костей и развитие скелета.
Синдром Шерешевского-Тёрнера лечится с помощью коррекции низкорослости путем проведения курсов человеческого соматропина, а также формирования вторичных половых признаков путем назначения эстрогенов.
Какой прогноз имеет синдром Шерешевского-Тёрнера?
Синдром Шерешевского-Тёрнера имеет благоприятный для жизни и интеллектуального развития прогноз. Неблагоприятный для деторождения.
Источник