
Синдром трипло х частота встречаемости

Синдром трисомии X представляет собой генетическое заболевание, наблюдаемое у женщин, характеризующееся наличием дополнительной Х-хромосомы.
Синдром Triple X также называют:
- 47, XXX
- 47, XXX Кариотип
- синдром XXX, 47
- Синдром XXX
- Трисомия X
Факты

- Встречается только у женщин
- Это не наследственное расстройство
- Синдром наблюдается у 1 из 1000 новорожденных девочек
- Некоторые случаи недиагностированы из-за отсутствия симптомов
- Приблизительно 10 процентов случаев диагностированы
Генетика синдрома Triple X
Обычно у каждого человека 46 хромосом, из которых две являются половыми, а именно X и Y. Женщины имеют две Х-хромосомы, мужчины одну Х и одну Y.
Люди, родившиеся с трисомией по х хромосоме, имеют 3 Х-хромосомы, поэтому общее количество составляет 47 из-за дополнительной Х.
Некоторые женщины с синдромом трисомии Х имеют дополнительную Х-хромосому только в некоторых клетках, которая называется 46, XX / 47, XXX мозаицизмом.
Причины и факторы риска
Трисомия по х хромосоме обычно не наследуется. Это происходит, когда репродуктивная клетка, имеет две Х-хромосомы из-за их нераспределения во время образования. Когда одна из таких клеток участвует в образовании зиготы, это приводит к синдрому тройного Х.
Причина мозаичной формы 46, XX / 47, XXX обусловлена аномальным делением клеток во время ранней эмбриональной стадии, что приводит к дополнительной Х-хромосоме только в некоторых клетках. Он также не является наследственным.
Симптомы и признаки
Симптомы и признаки синдрома трисомии Х широко варьируются среди пациентов. Пораженные женщины могут быть бессимптомными или иметь несколько симптомов или много аномалий. Ниже приведены аномалии, наблюдающиеся при трисомии по х хромосоме.
- Рост больше, чем средний, с длинными ногами.
- Задержка развития моторных навыков, таких как ходьба и сидение.
- Слабый мышечный тонус (гипотония).
- Низкий IQ: на 10-15 баллов ниже, чем у братьев, сестер.
- Отсроченные речевые и языковые навыки.
- Поведенческие и эмоциональные проблемы.
- Дефицит памяти, суждений, обработки информации.
- Маленькие пальцы или аномально кривые, называются клинодактильными.
- Младенцы с тройным X могут иметь эпикантальные складки (часть верхнего века, которая образует складку и покрывает внутренний угол глаза), гипертелоризм (увеличение пространства между двумя глазами), небольшая окружность головы.
- Тревожность.
- Гиперактивное расстройство с дефицитом внимания(ADHD): дети с СДВГ проявляют чрезмерную активность, отсутствие внимания, неконтролируемое поведение.
- Аномальное развитие яичников (дисвразия яичников).
- Раннее или отсроченное половое созревание.
- Преждевременная недостаточность яичников, бесплодие.
- Почечная агенезия (неспособность развиться) почечная дисплазия (аномальное развитие).
- Периодические инфекции мочевых путей.
- Плоскостопие.
- Запор, боль в животе.
- Pectus excavatum (аномальная грудная стенка, которая вогнута или утоплена)
- Сердечные аномалии.
Диагностика
Предполагается, что синдром трисомии Х возникает, когда пациент представляет любой из симптомов или задержку полового созревания или другие нарушения менструального цикла.
Хромосомный анализ
Анализ хромосом в клетках крови пострадавшего подтверждает диагноз в подозреваемых случаях.
Другие диагностические методы включают в себя пренатальную диагностику, которая выполняется у некоторых пациентов по другим причинам, а состояние диагностируется случайно.

Амниоцентез
У беременных женщин проводится проверка хромосомных аномалий растущего плода. Это инвазивная процедура. Амниотическая жидкость содержит клетки плода. Жидкость собирают и клетки исследуют, чтобы проверить количество хромосом и другие аномалии.
Если у плода есть синдром тройного Х, у клеток будет дополнительная Х-хромосома.
Выбор хорионических ворсинок
Выборка хорионических ворсинок (CVS) выполняется у беременных женщин, чтобы проверить хромосомные аномалии растущего плода. В плаценте присутствуют хорионические ворсинки. Собирают немного ткани плаценты и проверяют на аномалии хромосом. Если у плода есть трисомия по х хромосоме, у клеток будет дополнительная Х-хромосома.
Лечение
Лечение синдрома тройного Х зависит от возраста представления заболевания, его тяжести и симптомов.
Дети
Если новорожденному диагностирован синдром трисомии Х, ребенок должен оцениваться следующим образом:
- Первые 4 месяца: оценка развития мышечного тонуса и силы.
- До 12 месяцев: оценка языка, речи.
- В дошкольном возрасте: предварительная оценка ранних признаков проблем чтения.
- Для детей с синдромом тройного Х необходимо оценить функцию почек и сердца.
Детям с синдромом тройного Х, ранняя оценка и вмешательство дают отличный результат. Речевая, развивающая терапия, физиотерапия, консультирование являются ключевыми мерами вмешательства, когда это необходимо.
Лечение тревоги и СДВГ имеет важное значение при обнаружении.
Молодые девушки
Для девочек с синдромом тройного Х подростковый возраст может быть сложной фазой жизни. Для них требуется короткий период консультирования.
Женщины
Для женщин с бесплодием и нарушениями менструального цикла требуется тщательная проверка наличия первичной овариальной недостаточности.
Генетическое консультирование
Генетическая консультация среди пострадавших и членов их семей является полезной.
Профилактика
Синдром трисомии X не является предупреждаемым.

Часто задаваемые вопросы
- С каким специалистом надо консультироваться, чтобы исключить синдром Triple X?
В зависимости от возраста ребенка вам может потребоваться консультация педиатра или гинекологом по проблемам, связанным с половым созреванием. Если они будут подозревать синдром Triple X, вас направят к генетику для хромосомного анализа и кариотипирования.
Является ли он наследственным расстройством?
Тройной синдром X не наследуется.
Как можно его избежать
Синдром Triple X не является предупреждаемым. Ранняя диагностика и лечение имеют решающее значение для предотвращения долгосрочных последствий.
Понравилась статья? Поделись с друзьями:
Источник
Впервые синдром
трисомии по Х-хромосоме был описан П.
Джекобс с соавторами в 1959 году.
Они
обнаружили в ядрах эпителия слизистой
оболочки щеки больной два тельца полового
хроматина. В среднем женщины с кариотипом
ХХХ встречаются с частотой 1 – 1,4 на 1000
родившихся девочек. Кроме обычного
трисомного варианта 47, ХХХ у женщин
описаны полисомии по Х-хромосоме с
кариотипами 48, ХХХХ и 49, ХХХХХ.
Клиническая
картина этого заболевания чрезвычайно
разнообразна. Психиатр, эндокринолог
и гинеколог могут встретиться как с
отчетливыми клиническими проявлениями
этого синдрома, так и со стертыми формами.
Всем больным свойственно только
присутствие в кариотипе трех хромосом
Х. Около 30% таких больных сохраняют
генеративную функцию и имеют нормальных
детей.
Клинически
больные с трипло-Х имеют недоразвитые
яичники, гипоплазию матки, нерегулярный
менструальный цикл; у них рано наступает
вторичная аменорея или бывает
преждевременный климакс. У многих
больных обнаруживаются неспецифические
соматические дизморфии различной
выраженности; грубых аномалий развития
наружных половых органов не обнаружено.
Довольно часто у женщин с ХХХ-хромосомным
комплексом отмечается незначительное
снижение интеллекта в стадии дебильности.
Доказано, что среди них в несколько раз
чаще можно встретить лиц с психопатическими
чертами и наклонностью к расстройствам
шизофреноподобного круга. По данным
Филлипова Ю.И. (1971) у взрослых больных с
синдромом трипло-Х шизофрения протекает
неблагоприятно с выраженными изменениями
личности; они склонны к проявлению
эпилепсии, особенно в детском возрасте.
У таких больных частота трипло-Х в
несколько раз выше популяционных
показателей. На цитогенетическое
исследование больные чаще всего попадают
из психиатрических лечебниц и домов
инвалидов для детей, которые страдают
умственной отсталостью.
Многие
исследователи отмечают своеобразную
особенность: с увеличением числа Х-
хромосом в кариотипе до 4, 5 и более
клинические проявления синдрома
усиливаются. Больные, имеющие 4, 5 или
более Х-хромосом, умственны более
отсталые и, как правило, из-за эндокринного
дисбаланса у них резко нарушается
генеративная функция.
Предварительный
диагноз синдрома трипло-Х основан на
исследовании полового хроматина, Этот
метод позволяет различать больных с
аномальным комплексом Х-хромосом и
первичной эндокринной патологией.
Окончательный диагноз устанавливается
по результатам кариологического
исследования.
Больные с трипло-Х
могут иметь потомство.
Лечение в основном
симптоматическое и направлено на
коррекцию эндокринного дисбаланса, в
первую очередь на устранение нарушений
функции яичников.
Синдром Клайнфельтера.
Клинически
синдром Клайнфельтера описан в 1942 г., а
цитогенетически в 1959 г. Генетической
особенностью этого синдрома является
разнообразие цитогенетических вариантов
и их сочетания (мозаицизма). Обнаружено
несколько типов полисомии по хромосомам
Х и У у лиц мужского пола: 47, ХХУ; 48, ХХХУ;
49, ХХХХУ; 47, ХУУ; 48, ХУУУ; 48, ХХУУ; 49, ХХХУУ.
Наиболее распространен полисомный по
хромосоме Х синдром Клайнфельтера
(ХХУ). Общая частота его в популяции 1,2
случая на 1000 новорожденных. Примерно у
10% больных с синдромом Клайфельтера
наблюдается мозаицизм 46, ХУ/47, ХХУ.
Считают, что добавочная Х-хромосома в
60% случаев наследуется от матери.
Общеизвестно,
что с увеличением числа половых хромосом
в кариотипе больных в большей степени
проявляется задержка умственного
развития (встречается примерно у 25%
больных) и возникает ряд нетяжелых
пороков и микроаномалий (пороки сердца,
сколиоз, катаракта, изменение
дерматоглифических рисунков и т.д.). По
сравнению с аутосомными аномалиями
нарушения в системе половых хромосом
слабо влияют на фенотип. Это связано,
вероятнее всего с тем, что в организме
больного активна одна лишь Х-хромосома
(остальные инактивированы); псевдоаутосомный
участок Х-хромосомы значительно короче
любой из аутосом и к тому же число генов
в У-хромосоме невелико.
Основные
клинические проявления, как правило, у
таких больных проявляются в пре- и
пубертатном периоде. В период
новорожденности они не особенно
отличаются от своих сверстников: иногда
выявляется незначительная задержка
психомоторного развития и гипоплазия
яичек.
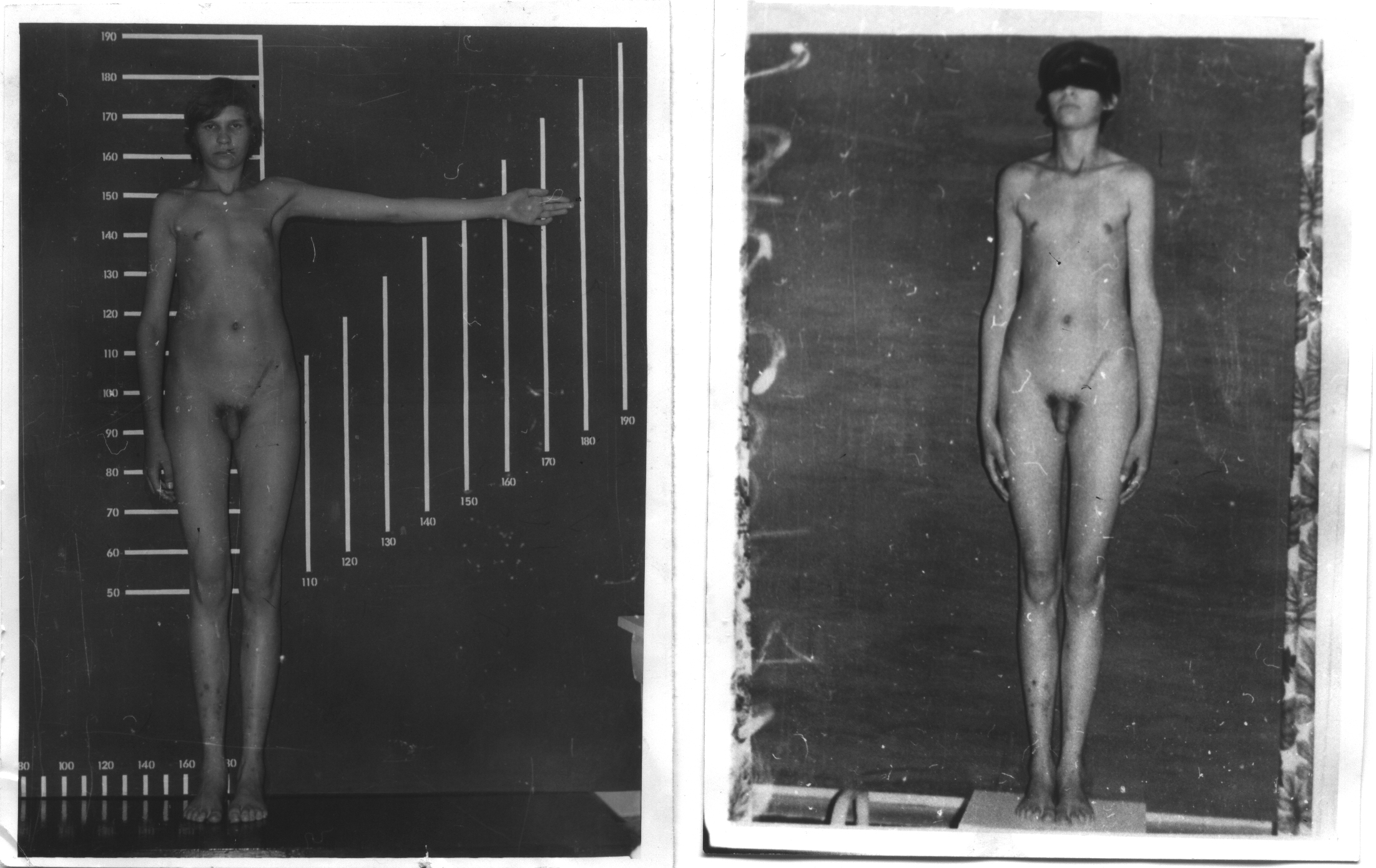
Для
мужчин с синдромом Клайнфельтера
характерны высокий рост, длинные
конечности, евнухоидизм и гинекомастия
(50%), нарушенный сперматогенез и в
результате этого бесплодие, уменьшенные
яички, повышенное выделение женских
половых гормонов, склонность к ожирению,
скудное оволосение в подмышечных
впадинах и на лобке.
4.5.8
Как
уже указывалось, лишняя Х-хромосома
обусловливает разнообразные нарушения
психики. Больные с этим синдромом очень
внушаемы, вялы, апатичны, у них часто
отмечается умственная отсталость
(обычно дебильность). В период полового
созревания повышается титр гонадотропинов
в моче; при электроэнцефалографическом
исследовании у некоторых больных
отмечают эпиактивность и различные
аномалии биоэлектрической активности
мозга. При патологоанатомическом и
гистологическом исследовании в яичках
обнаруживают гипоплазию, более или
менее выраженный гиалиноз и склерозирующую
дегенерацию семенных канальцев; в
гипофизе находят недостаток хромофобных
и избыток ацидофильных клеток.
Диагностировать
синдром Клайнфельтера, особенно у
взрослых лиц, нетрудно. Своеобразное
сочетание высокого роста, строения
скелета по женскому типу, гинекомастии,
ожирения и снижения интеллекта позволяет
даже без исследования полового хроматина
предполагать синдром Клайфельтера. При
определении в соскобе слизистой оболочки
щеки тельца полового хроматина и тем
более при кариотипировании лишней
Х-хромосомы диагноз этой болезни не
вызывает никаких сомнений.
Из
сопутствующих заболеваний у больных с
синдромом Клайнфельтера могут быть рак
молочной железы, сахарный диабет, болезни
щитовидной железы, хронические
обструктивные заболевания легких.
Повторный
риск рождения для синдрома Клайфельтера
не превышает общепопуляционные показатели
и составляет 1 случай на 2000 новорожденных.
Специфического
лечения нет; при симптоматической
терапии применяют гормональные препараты
(прогестерон, эстрадиола пропионат,
тестостерона пропионат и др.), которые
направлены на коррекцию вторичных
половых признаков. Однако пациенты даже
после терапии остаются бесплодными.
Психотерапия направлена на социальную
адаптацию таких больных в обществе.
Соседние файлы в предмете [НЕСОРТИРОВАННОЕ]
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
- #
Источник
Синдром нечувствительности к андрогенам ещё называют синдромом тестикулярной феминизации.
Относится к врождённым эндокринным нарушениями полового развития, вызванным перерождением гена, ответственного за рецептор андрогена.

Фенотипы людей с этим геном находятся в довольно широком диапазоне — от нормального телосложения, характерного для мужчины до практически полного женской комплекции, несмотря на то, что в их кариотипе присутствует Y-хромосома.
Заключение
- Люди с завершённым типом синдрома нечувствительности имеют внешность, больше всего напоминающую женскую: с грудью и влагалищем, не опустившимися яичками.
- При этом недуге нарушается соотношение мужских и женских гормонов.
- Причины заболевания — наследственные, обнаруживается в период внутриутробного развития.
- Болезнь относится к очень редким.
- Проявляется в виде феминизации.
- Диагностируется при УЗИ, осмотре, анализа крови и генотипа.
- Предупредить патологию практически невозможно, так как её появление зависит от случайных мутаций.

Что представляет собой синдром нечувствительности к андрогенам
Это заболевание характеризуется генетически обусловленным нарушением работы рецепторов, которые отвечают за реакцию на половые гормоны мужчин. Болезнь наследуется по линии матери (с Х-типом).
Если женщина является носителем мутированного гена, то риск возникновения феминизации повышается до 30 процентов.
До 6 — 7 недели половые органы эмбриона развивается без отличия того или иного типа. Когда андрогены доминируют, то плод формируется по мужскому образу. Если же таковых гормонов нет, то закладываются женские половые органы.
Подобные пациенты имеют мужской тип, поэтому и репродуктивная система формируется так же. Но из-за обусловленной вследствие мутаций нечувствительности рецепторов к андрогенам развитие ребёнка по мужскому типу останавливается.
После рождения у младенца постепенно проявляется синдром.
Причины и механизм развития синдрома тестикулярной феминизации
Главный механизм формирования синдрома Морриса — геномные расстройства в период внутриутробного созревания эмбриона. Из-за мутации генов, отвечающих за чувствительность рецепторов к специфическим мужским гормонам, плод начинает развиваться по женскому типу.
Несмотря на то что у него имеются в наличии Х и Y хромосомы (то есть он определяется к мужскому полу) из-за особенностей баланса гормонов и функционирования андрогеновых рецепторов первичные и вторичные мужские половые признаки не развиваются или же проявляются незначительно.
Все эти нарушения формируются в течение 1 триместра (до 12-й недели включительно).
Факторы, способствующие тестикулярной феминизации:
- сбои эндокринного баланса матери;
- срывы обмена веществ;
- употребление беременной алкоголя;
- курение во время вынашивания ребёнка;
- зависимость от наркотиков;
- применение неразрешённых медикаментов в период гестации;
- неправильная терапия гормонами.
Как можно распознать синдром Морриса у ребёнка
Существует абсолютная и частичная форма нечувствительности к андрогенам. Начальные формы тестикулярной нечувствительности определяются у юношей с начала пубертатного периода. Симптомы всегда своеобразны и зависят от типа патологии.
При абсолютной нечувствительности к андрогенам у лиц мужского пола наблюдается формирование тела и мочеполовых органов с ярко выраженной тенденцией феминизации. Присутствуют такие проявления:
- увеличение груди;
- практически полное отсутствие волос на лобке и в подмышечных впадинах;
- значительное усиление волос на голове;
- большие бёдра, маленькие плечи и другие нетипичные для мужского силуэта черты;
- инфантилизм (недоразвитие) половых губ, репродуктивных органов.
Неполная форма синдрома имеет более разнообразную клиническую картину.
Как диагностируется синдром резистентности к андогенам
Методы диагностики этого недуга:
- осмотр гинеколога или уролога;
- анамнез;
- определение гормонов.
Ранние конфигурации тестикулярной феминизации заметны уже при осмотре неонатолога. Неполная форма по мужскому типу диагностируется намного позже. Предлогом для диагностики часто служит обращение мужчины к врачу по поводу бесплодия. При исследовании спермы обнаруживается азооспермия.
Анализ на тестостерон часто показывает значительно увеличение концентрации этого гормона в крови.
Нужно ли лечить это заболевание
Необходимо лечить все формы данной патологии. Показаны хирургическое вмешательство и коррекция гормонального фона.
Прогноз заболевания
У женщин и мужчин с наличием синдрома нечувствительности к андрогенам в большинстве случаев полностью отсутствуют половые железы. Вследствие этого, они теряют возможность зачать и родить ребёнка, так как являются бесплодными. В данном случае выход один — усыновление.
Частота встречаемости и виды заболевания
Болезнь встречается очень редко: на 1 млн жителей бывает не больше 20 случаев патологии.
Выделяют 5 степеней неполной нечувствительности.
- Мужской тип. Выраженных отклонений не наблюдается. В крайне редких происшествиях у пациентов отмечается слишком высокий голос. Процессы образования сперматозоидов нарушаются, почти всегда мужчины страдают бесплодием.
- Феминизация по преимущественно мужскому типу. Фенотипически пациенты относятся к мужчинам. При диагностике андрогенной нечувствительности обнаруживается гипоспадия (расщепление уретры), выраженное уменьшение полового члена, неравномерное разделение подкожного жира.
- Амбивалентный тип нечувствительности к андрогенам характеризуется значительным снижением размера пениса, делением мошонки (она больше напоминает половые губы), неопущением яичек. У больных расширен таз, сужены плечи.
- Феминизация по преимущественно женскому типу отличается тем, что пациент фенотипически является женщиной. Характерно значительное увеличение клитора. Нередко половые губы у таких представительниц сращены.
- Женская форма андрогенной нечувствительности характерен тем, что у больных симптомов вирилизации практически нет. Возможно частое развитие пиелонефрита и уретрита.
Осложнения и последствия
Течение болезни не представляет собой угрозы для жизни. Но из-за изменения внешнего вида гениталий и бесплодия пациенты подавлены, склонны к частым депрессиям.
Наиболее грозное осложнение патологии — рак яичек, семинома. Данная аномалия формируется в результате злокачественного перерождения тестикула. Хирургическая операция помогает не допустить развития недуга.
Лечебные мероприятия
Лечение синдрома нечувствительности к андрогенам полностью зависит от формы болезни.
Гормональная заместительная терапия
В комплекс лечебных мероприятий входит коррекция гормонального фона (при необходимости).
Психологическая поддержка
Больные при андрогенной нечувствительности обычно бесплодны, они часто нуждаются в психологической помощи.
Пластическая хирургия
Лечение полного типа нечувствительности к андрогенам ограничивается удалением мужских половых желёз. Удаление яичек необходимо для недопущения развития семиномы.
Лечение неполной формы нечувствительности часто может быть связано с большим количеством оперативных вмешательств для создания нормальной формы гениталий и коррекции работы мочевыделительной системы.
Как выглядят пациенты с неполной формой синдрома Морриса
На этих фото изображены больные с разными формами нечувствительности к андрогенам.
Таблица симптомов 5 различных форм неполного синдрома
В этой таблице обобщённо признаки неполной формы синдрома нечувствительности к андрогенам.
Диагностика синдрома резистентности к андрогенам
Иногда опытный доктор может установить диагноз уже в ходе медицинского осмотра. Подтверждают его генетический анализ и развёрнутое исследование крови.
Дифференциальный диагноз
Нужно дифференцировать нечувствительность к андрогенам от:
- болезни Рокитанского-Кюстнера (врождённая аномалия, которая характеризуется отсутствием матки и верхних отделов вагины);
- чистой формы дискинезии гонад;
- гермафродитизма;
- синдрома Кляйнфельтера;
- дисфункции коры надпочечников.
Образ жизни пациентов
При коррекции формы гениталий и соответствующей гормонотерапии больные способны вести половую жизнь. Иногда внешний вид мужчины с нечувствительностью ничем не отличается от здоровых представителей сильного пола.
Фертильность у таких лиц исключена.
Отзывы
Анна, г. Москва
«У сына диагностировали неполную нечувствительность к андрогенам. Проводим гормональное лечение, пубертатное развитие проходит нормально».
Елена, г. Волгоград
«У меня диагностирована неполная нечувствительность к андрогенам. Проведена коррекция влагалища, принимаю гормоны. Надеюсь, что процедура искусственного оплодотворения протечёт успешно».
Оксана, г. Санкт-Петербург
«Гормональное лечение андрогенной нечувствительности у ребёнка миновало неудачно. Поэтому записались в очередь на хирургическое удаление семенников и пластическую операцию».
Источник