
Синдромом хатчинсона гилфорда детская прогерия что это

Прогерия – редкое генетическое заболевание, характеризующееся преждевременным старением организма, соответствующими изменениями внутренних органов. Проявляется гиперпигментацией, истончением и утратой эластичности кожи, поседением и выпадением волос, увеличением размеров черепа, уменьшением его лицевой части, экзофтальмом, развитием атеросклероза сосудов, инфаркта и фиброза миокарда, остеопороза, сахарного диабета, формированием злокачественных опухолей. Диагностика основана на сборе клинических данных, лабораторном исследовании ДНК. Лечение направлено на устранение симптомов заболевания: профилактику и терапию атеросклероза, инфаркта миокарда, диабета, остеопороза, рака.
Общие сведения
Термин «прогерия» в переводе с древнегреческого языка означает «преждевременное старение». Детская форма прогерии называется синдромом Хатчинсона-Гилфорда согласно фамилиям исследователей, которые впервые независимо друг от друга описали данную патологию: британский врач Дж. Хатчинсон – в 1889 году, его соотечественник Х. Гилфорд – в 1897 году. Взрослая форма носит название «синдром Вернера», так как в 1904 году немецкий врач О. Вернер первым официально наблюдал симптомы старения у подростков 14-19 лет. Оба варианта болезни являются крайне редкими. Распространенность детской прогерии составляет 1 случай на 7-8 млн. человек, с момента начала исследований патологии выявлено около 150 больных детей. Взрослая прогерия встречается чаще, средние эпидемиологические показатели – 1 больной на 100 тыс. населения.
Прогерия
Причины прогерии
В основе заболевания лежит генетическая мутация. Причиной детского типа прогерии является дефект гена LMNA, который кодирует особый белок ламин A, выстраивающий оболочку ядра клетки. Как правило, мутация происходит спорадически – во время созревания половых клеток у родителей или при зачатии. Однако исследователи выявили несколько случаев патологии у родных братьев и сестер, часть из которых входила в семьи с близкородственными браками. Это свидетельствует о возможности наследования мутации по аутосомно-рецессивному типу (родители являются носителями дефектного гена, но не болеют и способны к деторождению).
Прогерия взрослых – наследственное заболевание. Оно развивается при наличии дефекта в гене RECQL2, который ответственен за производство белка WRN, поддерживающего стабильность генома, целостность и структуру ДНК. Передача мутации происходит аутосомно-рецессивным способом, болезнь проявляется у людей, получивших два дефектных гена в одной аллели (от матери и от отца).
Патогенез
При детском варианте прогерии генетический дефект приводит к нарушению синтеза структурно правильного белка ламина А, образующего внутренний каркас ядерной оболочки. В результате мутации налаживается производство укороченного белка-предшественника преламина А, а строение созревшего конечного белка отличается от нормального. Молекулы белка встраиваются в ядерную мембрану, изменяют ее форму и повышают хрупкость. Ядро не может сохранять свою целостность, клетка гибнет раньше времени.
При болезни Вернера возникает недостаточность WRN-белка, определяется генетически детерминированная нестабильность хромосом. Их структура очень часто изменяется спонтанно или под воздействием некоторых факторов. В итоге ухудшается способность клеток к делению, пролиферативный потенциал становится в 3-5 раз меньше, чем должен быть в норме. В 10 раз увеличивается частота спонтанных мутаций, аномально укорачиваются теломеры – концевые участки хромосом, защищающие гены от повреждений.
Симптомы прогерии
Прогерия Хатчинсона дебютирует после периода нормального развития в возрасте от 6 месяцев до 2 лет. Изменяется внешность детей: замедляется рост, увеличиваются размеры черепа, но лицевая часть остается маленькой, нижняя челюсть – недоразвитой. Формируется клювовидный тонкий нос и экзофтальм (выпячивание глаз). Тотально выпадают волосы – на голове и теле, ресницы и брови. При алопеции разрушаются волосяные фолликулы, поэтому дальнейший рост волос становится невозможным. На волосистой части головы выбухают вены. Дерма и подкожная клетчатка подвергаются атрофическим изменениям: кожа истончается, иссушается, покрывается морщинами. Развивается липодистрофия – заметное снижение количества подкожного жира. Относительно сохранным он остается на щеках и лобке.
На туловище образуются склеродермоподобные очаги – уплотнения в нижней части живота, на ягодицах и бедрах. На открытых участках кожи появляются пигментные пятна. Ногти недоразвитые, ломкие, утолщенные, округло выпуклые – имеют форму «часовых стекол». К 2 годам развивается фиброз органов и околосуставных тканей. Пассивные движения в локтевых и коленных суставах ограничены (контрактуры), формируется характерное положение костей ног – «поза всадника». Скелет подвергается гипоплазии, дисплазии и дегенеративным изменениям. Молочные и постоянные зубы прорезываются с опозданием, частично отсутствуют, скученные, искривленные, подвержены кариесу. К 5-6 годам диагностируется атеросклероз сосудов, сердечные шумы, гипертрофия левого желудочка, помутнение хрусталика, резистентность к инсулину. Половые органы остаются недоразвитыми. Уровень умственного развития обычно выше, чем у сверстников.
Клинические проявления синдрома Вернера обнаруживаются с 14 до 18 лет. Подросток начинает отставать в росте, его волосы седеют и выпадают. К 20 годам пациенты лысеют. Кожа лица и конечностей бледнеет, истончается, натягивается. Под ней видна сеть кровеносных сосудов. Мышечная и жировая ткани атрофируются, руки и ноги становятся непропорционально тонкими, кожа над суставными выступами изъязвляется. К 30 годам появляется катаракта, голос слабеет и хрипнет, на ногах образуются язвы, на подошвах – мозоли, сосудистые звездочки, кератоз. Внешний вид пациентов специфичен: низкий рост, лунообразное лицо, выступающий подбородок, суженное ротовое отверстие, псевдоэкзофтальм.
Сальные и потовые железы атрофируются. Костно-суставные изменения включают метастатическую кальцификацию, явления генерализованного остеопороза, эрозивных остеоартритов, ограниченную подвижность и деформацию пальцев рук, сгибательные контрактуры, боли в конечностях, артрит и остеомиелит. Отмечаются атеросклеротические изменения сосудов, медленно прогрессирует катаракта, снижаются интеллектуальные способности. После 30 лет проявляются эндокринные заболевания – сахарный диабет, гипогонадизм, тиреопатии. У 5-10% больных обнаруживаются злокачественные опухоли различных органов, костей и кожи. Причиной смерти становится онкопатология или тяжелое сердечно-сосудистое заболевание.
Диагностика
Диагноз прогерии устанавливается на основании клинико-анамнестических данных. В зависимости от имеющейся симптоматики в диагностике могут принимать участие эндокринологи, неврологи, дерматологи, терапевты, врачи-генетики. Детскую и взрослую прогерию необходимо дифференцировать с системной склеродермией, пойкилодермией, синдромом Ротмунда-Томсона, синдромом Видемана-Раутенштрауха. К основным методам обследования больных относятся:
- Опрос. В ходе беседы врачи уточняют время появления симптомов, их выраженность, степень физической и социальной дезадаптации, наличие периода обычного развития (6-24 месяца при детской форме, 14-20 лет – при взрослой). Собирают данные семейного и генеалогического анамнеза, выявляя генетическую патологию или ее отсутствие в роду.
- Осмотр. Специалисты оценивают неврологический статус, пассивную и активную подвижность суставов, костные деформации, состояние кожи и подкожной клетчатки, сохранность зрения, интеллектуальных функций. Пациенты могут быть направлены на инструментальные исследования: рентгенографию костей, УЗИ и МР-томографии органов, ЭКГ, офтальмоскопию.
- Биогенетическое исследование. Выполняется анализ генетического материала методом секвенирования ДНК. Исследуются участки генов, дефекты в которых приводят к развитию прогерии. У пациентов с детской и взрослой формой болезни обнаруживаются мутации в гомозиготном и компаунд-гетерозиготном состоянии.
Лечение прогерии
Специфические методы терапии прогерии не разработаны. Медицинская помощь пациентам заключается в облегчении симптомов болезни. Проводится лечение атеросклероза, остеопороза, остеомиелита, катаракты, диабета, патологий сердца и онкологических заболеваний. Известен случай, когда состояние больного заметно улучшилось на несколько лет после операции трансплантации сердца.
Научные исследования ведутся в области генной инженерии – совершаются попытки выделить мутационный ген из ДНК и заменить его здоровым. Параллельно разрабатывается метод этиотропной терапии. Ученые из Швеции нашли способ нейтрализации дефектных молекул ламина A, которые проникают в мембрану клеточных ядер и способствуют гибели клетки. Положительные результаты получены на экспериментах с мышечной тканью крыс, на людях препарат пока не опробован. В США с 2012 года ведется изучение эффективности применения ингибитора фарнезилтрансферазы (лекарственного средства, разработанного для лечения рака). После терапии дети из экспериментальной группы чувствовали себя лучше, повысилась жесткость и эластичность артерий, плотность костей, снизились случаи транзиторных ишемических атак, сердечных патологий, утраты зрения и слуха. Продолжительность жизни увеличилась на 1,6-6 лет.
Прогноз и профилактика
В среднем больные детской прогерией доживают до 13 лет, зарегистрированы случаи смерти в 7 и 27 лет. Продолжительность жизни при взрослом типе заболевания составляет 30-40 лет. Причиной летального исхода становятся атеросклеротические осложнения и злокачественные новообразования. Профилактические меры отсутствуют. Предупредить рождение больного ребенка можно при наследственном характере болезни – при синдроме Вернера, в отдельных случаях синдрома Хатчинсона, когда патология выявлена в семье, будущие родители отнесены к группе высокого риска. Таким парам назначается медико-генетическое консультирование.
Источник
Общие сведения
Прогерия – это генетическое заболевание, которое считается одним из редчайших, за всю историю человечества зафиксировано примерно 350 случаев. Самыми известными людьми с прогерией считаются диджей Леон Бота и мотиватор Сэм Бернс. Прогерия в России не очень распространена – зарегистрировано всего около 80 случаев.
Главной особенностью этого генетического дефекта является преждевременное старение организма, которое сопровождается существенными изменениями кожных покровов и работы внутренних органов. С греческого progeros переводится как «преждевременно постаревший».

Леон Бота
Как указывает Википедия заболеванию присвоен код МКБ-10: Е34.8 и относится к другим уточненным эндокринным расстройствам.
Патогенез
В основе патогенеза детской прогерии – внезапная мутация в гене LMNA (в единичных случаях бывает врожденная), ген отвечает за кодировку ламин А – белков, встроенных в ядерную ламину и удерживающих оболочки ядра клеток. Ядерная ламина – жесткая структура из фибриллярной сетки, связанная с нитями хроматина, участвующая в их организации.
 Нарушения этих структур приводит к нестабильности ядер клеток, нарушению процессов репарации ДНК, клонирования фибробластов, что вызывает старение всего организма и запускает такие классические процессы в результате деградации клеточного кода, как облысение и поседение, нарушения работы сердечно-сосудистой системы, ухудшение зрения, генерализованный остеопороз, онкообразование.
Нарушения этих структур приводит к нестабильности ядер клеток, нарушению процессов репарации ДНК, клонирования фибробластов, что вызывает старение всего организма и запускает такие классические процессы в результате деградации клеточного кода, как облысение и поседение, нарушения работы сердечно-сосудистой системы, ухудшение зрения, генерализованный остеопороз, онкообразование.
Классификация
Различают детскую прогерию — синдром Хатчинсона Гилфорда и взрослую — синдром Вернера.
В отличие от детской, проявляющейся в 5-8-месячном возрасте, у взрослых симптомокомплекс преждевременного старения развивается примерно в 20-30 лет, начинаясь в период полового развития. Тело и лицо начинает меняться как на картинке ниже (прогерия — фото) с разницей в 33 года: приобретает заостренные черты, нос становится похожим на птичий, рот узким, голос высоким и хриплым, кожа бледной, конечности тонкими, нет подкожной клетчатки. При этом происходит атрофия мышц, образование трофических язв, гипогонадизм, прогрессия глаукомы, катаракты, атеросклероза, облысения и поседения, выпадение зубов, снижение интеллекта.

Синдром Вернера, пациентке 15 и 48 лет
Прогерия: причины заболевания
Заболевание наследственное и передается по аутосомно-рецессивному типу, поэтому единичные случаи братьев и сестер с прогерией объясняются тем, что родители были достаточно близкими родственниками, например, кузенами и заболевание стало результатом кровосмешения. Однако, называют и другие причины заболевания, например:
- диэнцефально-гипофизарная недостаточность;
- вторичное поражение нескольких желез эндокринной системы;
- является результатом одного из проявлений других эндокринных наследственных болезней.
Также предполагается, что синдром Хатчинсона Гилфорда возникает в результате спорадической – внезапной мутации в сперматозоиде или яйцеклетке еще до момента зачатия.
Симптомы
Как синдром преждевременного старения, болезнь прогерия вовлекает в патогенетический процесс различные системы и органы:
- Изменения кожных покровов и волос: сухая, морщинистая (пергаментная) кожа, редкие седые волосы, отсутствие бровей и ресниц, истончение и ломкость ногтевых пластинок.
- Изменения черепа: непропорционально телу большая голова, лицо маскообразное с крючковатым носом, уши оттопыренные, глаза экзофтальмированные, зубы отсутствуют либо прорезаются поздно.
- Изменения костно-мышечного аппарата: слаборазвитая мускулатура, низкий рост, конечности тонкие, остеопороз, постоянное ощущение скованности, отсутствует подкожная жировая прослойка в результате стремительной потери веса и остановки роста.
- Половая система: недоразвита в результате гипоплазии половых органов, нет вторичных половых признаков.
- Наиболее существенные и опасные симптомы происходят со стороны сердечно-сосудистой системы: развивается генерализированный атеросклероз, что приводит к тромбозу коронарных артерий и инфарктам миокарда.
- Неврологические нарушения: снижение интеллекта.
- Прочее: склерозирование и нарушение работы головного мозга, печени, почек, эндокринных органов в результате отложения жироподобных веществ.
Анализы и диагностика
Благодаря работе исследовательских фондов изучения прогерии на сегодняшний день есть возможность идентифицировать генные мутации программами диагностического тестирования и помочь больным на более ранних стадиях развития заболевания.
Чтобы узнать есть ли генетические отклонения у ребенка достаточно посетить квалифицированного врача, который изучит внешний вид, данные анамнеза и направит на анализ крови, выявляющий генетические дефекты.
Лечение
Помощь особам, подверженным преждевременному старению должна быть комплексной, включая симптоматическое лечение, а также:
- непрерывный уход;
- кардиологическая помощь;
- специальное питание;
- физическая терапия.
Ученые выдвигают предположение, что ингибиторы фарнезилтрансферазы (FTIs), которые помогают в борьбе с раком, также способны обратить процессы образования структурных аномалий.
Обычная медикаментозная терапия включает прием:
- Аспирина – в малых дозах ацетилсалициловая кислота помогает «разжижать» кровь и препятствовать инфарктам и инсультам.
- Гормоны роста – гормональная заместительная терапия помогает решать проблему остановки роста и стимулирует процессы наборы веса.
Процедуры и операции
- Стоматологическая помощь для формирования нормального зубного ряда.
- Трудотерапия и физические нагрузки для снижения скорости формирования тугоподвижности суставов.
- При генерализированном атеросклерозе могут понадобиться хирургические вмешательства — коронарное шунтирование или стентирование для снижение риска инфаркта.
Детская прогерия
Детская прогерия начинается внезапно в 5-8 месяцев либо реже в 3-4 года и проявляется в виде резкого замедления темпов физического развития.
При таком страшном летальном диагнозе очень важно выявить недуг на ранних стадиях, когда в организме еще не произошли необратимые процессы. Не смотря на то, что лекарство еще не изобрели, очень важно ребенку своевременно получить симптоматическое лечение, правильно выстроить его режим и питание с учетом особенностей организма.
Диета при прогерии
Питание для предотвращения преждевременного старения должно быть калорийно ограниченным и качественно полноценным. Помимо этого диета должна быть:
- богата антиоксидантами и включать витамины С, Е, рутин и пр.;
- с регулярными мероприятиями энтеросорбции для обезвреживания токсических вещества, например, прием активированного угля;
- в рационе должны каждый день присутствовать кисломолочные продукты;
- нужно ограничить потребление жареного красного мяса, алкоголя, белого сахара и мучных продуктов.
Прогноз
Считается крайне неблагоприятным, так как единой стратегии лечения до сих пор не разработано и продолжительность жизни не превышает 27 лет, в среднем составляет 13 лет. Причинами летального исхода чаще всего становится истощение, инфаркт миокарда, злокачественные новообразования, интеркуррентные заболевания – случайно присоединившиеся и осложняющие течение самой прогерии. Особы с взрослой прогерией обычно доживают до 40-50 лет.
Список источников
- Козлова С. И., Демикова Н. С., Семанова Е. и др. Наследственные синдромы и медико-генетическое консультирование, 1996. – 242 с.
- Висмонт Ф.И., Леонова Е.В., Чантурия А.В. Общая патофизиология. – Минск: Выш. шк., 2011. – 91 с.
Источник
Общая информация
Прогерия, или синдром Хатчинсона-Гилфорда или синдром преждевременного старения, является врожденным, редким, смертельным генетическим заболеванием с поразительными чертами, напоминающими преждевременное старение.
Дети с прогерией обычно имеют нормальный вид в раннем детстве. В возрасте примерно от 9 до 24 месяцев у детей, подверженных заболеванию, начинаются глубокие задержки роста, что приводит к низкому росту и низкому весу. У них также появляются изменяется черты лица, характеризующиеся:
- непропорционально маленьким лицом по сравнению с головой;
- недоразвитая челюсть (микрогнатия);
- пороки развития и скученность зубов;
- выразительные глаза;
- маленький носик;
- тонкая синева вокруг рта.
Кроме того, ко второму году жизни у людей с синдромом преждевременного старения волосы на голове, брови и ресницы выпадают (алопеция), и волосы на голове могут быть заметно маленькими, белыми или светлыми. Дополнительные характерные признаки включают генерализованный атеросклероз, сердечно-сосудистые заболевания и инсульт, вывихи бедра, необычно заметные вены кожи головы, потерю слоя жира под кожей (подкожная жировая ткань), дефекты ногтей, скованность суставов, дефекты скелета и/или другие нарушения.
Согласно сообщениям в медицинской литературе, у людей с синдромом Хатчинсона-Гилфорда развивается преждевременное, широко распространенное утолщение и потеря эластичности стенок артерий (атеросклероз), что приводит к опасным для жизни осложнениям в детстве, подростковом или раннем взрослом возрасте.
Дети с прогерией умирают от болезней сердца (атеросклероза) в среднем в возрасте 14 лет с диапазоном от 8 до 21 года. Как и с любым человеком, страдающим сердечно-сосудистыми заболеваниями,
Синдром преждевременного старения вызван мутацией гена LMNA или ламина A. Белок ламина A представляет собой каркас, удерживающий вмести ядро клетки. Ученные считают, что дефектный белок ламина А, делает ядро нестабильным. Эта клеточная нестабильность, по-видимому, приводит к процессу преждевременного старения при прогерии.
Признаки и симптомы прогерии

У новорожденных с синдромом Хатчинсона-Гилфорда могут быть некоторые подозрительные признаки, присутствующие при рождении, такие как:
- необычно тугая, блестящая, твердая (т.е., «склеродермоподобная») кожа над ягодицами, верхними ногами и нижней частью живота;
- синеватое изменение цвета кожи и слизистых оболочек в средней части лица (срединно-лицевой цианоз);
- «скульптурный» нос.
Глубокая прогрессирующая задержка роста обычно проявляется примерно к 24 месяцам, что приводит к низкому росту и весу.
Согласно сообщениям в медицинской литературе, дети в возрасте 10 лет, как правило, имеют рост, близкий к росту среднего трехлетнего ребенка.
Ко второму году жизни также наблюдается недоразвитие (гипоплазия) костей лица и нижней челюсти (микрогнатия). Лицо кажется непропорционально маленьким по сравнению с головой, а кости спереди и по бокам черепа необычно заметны (лобное и теменное выпуклость).
У больных детей обычно есть дополнительные характерные черты лица, в том числе:
- маленький, тонкий, потенциально заостренный нос;
- необычно заметные глаза;
- маленькие уши с отсутствующими долями;
- тонкие губы.
Также могут присутствовать зубные аномалии, такие как:
- отсроченное прорезывание первичных (молочных) и вторичных (постоянных) зубов;
- неправильно сформированные, маленькие, обесцвеченные зубы;
- необычно повышенная частота кариеса.
Кроме того, ненормальная малость челюсти может привести к скученности зубов.
У детей с синдромом преждевременного старения волосы на голове обычно теряются примерно к двум годам. Волосы на голове могут быть тонкими, белыми или светлыми, в некоторых случаях такими могут сохраняться на протяжении всей жизни. Кроме того, брови и ресницы также могут быть потеряны в раннем детстве.
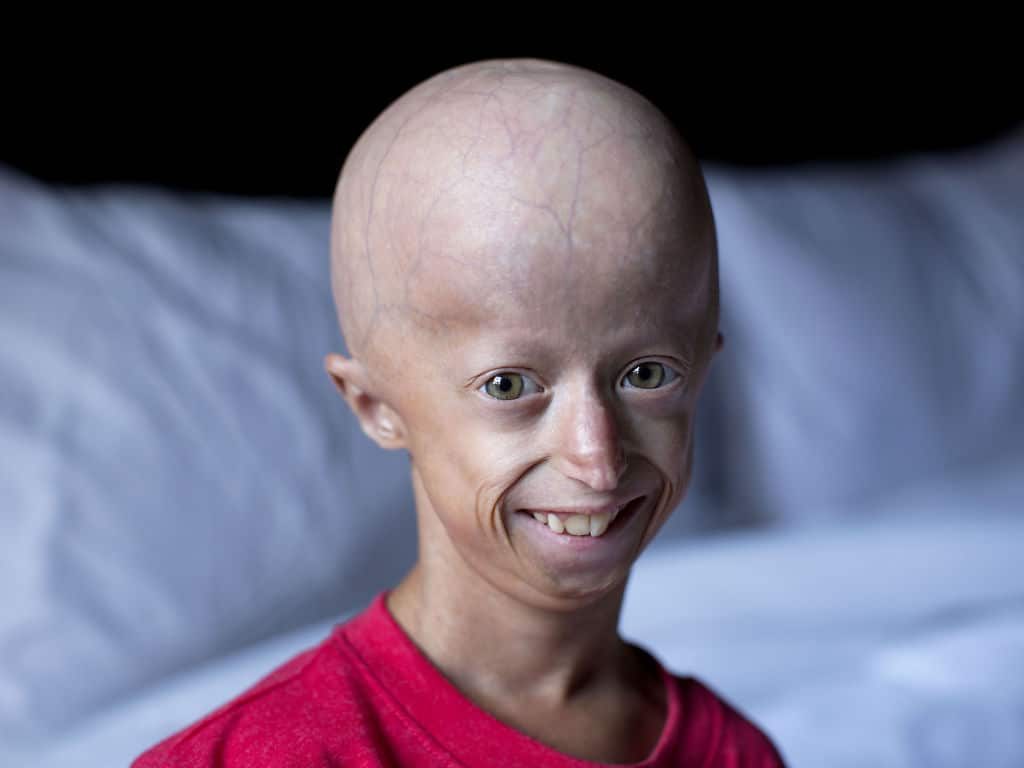
Синдром Хатчинсона-Гилфорда также характеризуется выраженными кожными нарушениями. Как обсуждалось выше, у новорожденных с таким расстройством могут быть «склеродермоподобные» изменения кожи на ягодицах, бедрах и нижней части живота. Кроме того, начиная с младенчества, происходит постепенная, почти полная потеря подкожной жировой ткани, и вены в определенных областях тела, особенно над кожей головы и бедрами, становятся ненормально заметными.
У больных детей кожа приобретает аномально состарившийся вид с необычно тонкими, сухими, морщинистыми и/или необычно блестящими и подтянутыми участками. Кроме того, с возрастом на участках кожи, подверженных воздействию солнца, могут появляться коричневатые кожные пятна. Больные дети обычно имеют дефекты ногтей, например, желтоватые, тонкие, хрупкие, изогнутые ногти или их полное отсутствие.
Дети с прогерией также имеют характерные дефекты скелета. К ним может относиться запоздалое закрытие «мягкого пятна» в передней части черепа (передняя родничок), аномально тонкой «куполообразной» части черепа и/или отсутствие определенных заполненных воздухом полостей внутри черепа, открывающийся в нос (околоносовые или лобные пазухи). Во многих случаях у детей также имеются:
- короткие, тонкие ключицы;
- узкие плечи;
- тонкие ребра;
- узкая или «грушевидная» грудь с выступающим животом.
Кроме того, длинные кости рук и ног могут казаться необычайно тонкими и хрупкими и склонны к переломам, особенно кости верхних рук (плечевые кости).
У многих детей с нарушениями скелета возникают дегенеративные изменения (остеолиз), которые могут поражать:
- ключицы;
- кости на концах пальцев (терминальные фаланги), в результате чего пальцы выглядят необычно короткими и «конусообразными»;
- тазобедренный сустав (вертлужная впадина).
Дегенеративные изменения в тазобедренном суставе могут привести к деформации тазобедренного сустава, при которой наблюдается аномальное увеличение угла кости бедра, боль в бедре и вывих бедра.
Кроме того, у многих больных детей вокруг определенных суставов (кисти, ступни, колени, локти и позвоночник) постепенно образуются аномальные фиброзные ткани (периартикулярный фиброз), вызывая необычную выпуклость, жесткость и ограниченное движение пораженных суставов.
Из-за скованности коленей, прогрессирующей деформации бедра, и другими связанными с мышечно-скелетными нарушениями дети с расстройством обычно имеют характерную, широко распространенную «стойку всадника» и шаркающую манеру ходьбы. Прогерия также связана с общей потерей плотности кости (остеопорозом), состоянием, вызывающим или способствующий повторным переломам после незначительной травмы.
Дополнительные симптомы и признака связанные с синдромом Хатчинсона-Гилфорда могут включать:
- сильно выделяющийся высокий голос;
- отсутствие груди или соска;
- отсутствие полового созревания;
- нарушение слуха и и прочие нарушения.
Согласно сообщениям в медицинской литературе, у пораженных детей в возрасте до 5 лет может развиться широко распространенное утолщение и потеря эластичности стенок артерий (атеросклероз). Такие изменения могут быть наиболее очевидными в определенных кровеносных сосудах, например, артериях, транспортирующих богатую кислородом кровь к сердечной мышце (коронарные артерии) и главной артерии тела (аорта).
Дополнительные признаки могут включать увеличение сердца (кардиомегалия) и аномальные шумы в сердце. В детском или подростковом возрасте прогрессирующий артериосклероз может привести к:
- периодическим болям в груди из-за недостаточного снабжения сердечной мышцей кислородом;
- затруднению кровотока в кровеносных сосудах головного мозга (цереброваскулярная окклюзия);
- прогрессирующей неспособности сердца эффективно перекачивать кровь в легкие и остальную часть тела (сердечная недостаточность);
- локализованной гибели части сердечной мышцы, вызванной прекращением ее кровоснабжения (инфаркт миокарда или сердечный приступ).
Причины прогерии
Ученные установили, что причиной возникновения прогерии является однобуквенная ошибка в гене на хромосоме 1, которая кодирует ламин A, белок, являющийся ключевым компонентом мембраны, окружающей ядро клетки. Аномальный белок ламина А, продуцируемый при синдроме, называется прогерин.
Прогерия обычно не передается по наследству. Изменения в генах — почти всегда случайное явление, встречающиеся крайне редко. Дети с другими типами прогероидных синдромов, которые не болеют синдромом Хатчинсона-Гилфорда, могут иметь заболевания, передающиеся по наследству.
Тем не менее, прогерия вызвана спорадической аутосомно-доминантной мутацией — спорадической, потому что это новое изменение в гене, и доминантной, потому что для того, чтобы синдром возник, необходимо изменение только одной копии гена. Для родителей, у которых никогда не было ребенка с прогерией, шансы родить ребенка с болезнью составляют 1 к 4-8 миллионам. Но для родителей, у которых уже есть ребенок с прогерией, шансы повторного рождения ребенка с заболеванием, намного выше — около 2-3%. Это связано с состоянием, называемым мозаицизмом, когда родитель имеет генетическую мутацию болезни в небольшой части своих клеток, но сам не болеет.
Конкретная первопричина ускоренного старения, связанная с синдромом Хатчинсона-Гилфорда, пока не известна. Многие ученные предполагают, что ненормальный процесс старения происходит из-за кумулятивного повреждения клеток в результате продолжающихся химических (метаболических) процессов в клетках организма.
Согласно этой теории, во время химических реакций, в организме образуются определенные соединения, называемые свободными радикалами. Считается, что увеличивающееся накопление свободных радикалов в тканях организма в конечном итоге приводит к повреждению и нарушению функционирования клеток, что в конечном итоге и приводит к старению. Считается, что некоторые ферменты (известные как антиоксидантные) играют роль в сдерживании процесса старения, способствуя устранению вредных свободных радикалов.
Ферменты — это белки, вырабатываемые клетками, ускоряющие скорость химических реакций в организме. Некоторые ученные указывают, что снижение активности некоторых ферментов может играть роль в ускоренном старении у людей с синдромом Хатчинсона-Гилфорда.
В одном исследовании клетки кожи (фибробласты), полученные от людей с прогерией, сравнивали с клетками кожи людей без заболевания. В фибробластах пациентов с прогерией уровни активности некоторых первичных антиоксидантных ферментов (например, глутатионпероксидазы [GPx], каталазы [CAT]) были значительно ниже, чем уровни, присутствующие в здоровых фибробластах.
Исследования показали, что прогерин вырабатывается гораздо более низкими уровнями у здоровых людей, и обычно накапливается в коронарных артериях в течение жизни с возрастом. Это открытие подтверждает возможность того, что прогерин является фактором риска развития атеросклероза среди населения земли в целом, и заслуживает изучения как потенциальной новый веток, помогающей прогнозировать риск сердечно-сосудистых заболеваний.
Таким образом, ученные подтвердили связь между нормальным старением, болезнями сердца и прогерией, поэтому поиск лекарства поможет не только больным детям, но, возможно, и тем, кто страдает от сердечных приступов, инсультов и других связанных со старением состояний.
Затронутые группы населения
Прогерия — редкое заболевание, одинаково влияющая на мужчин и женщин, и все расы. Это расстройство было первоначально описано в медицинской литературе в 1886 г. (Джонатан Хатчинсон) и 1897 г. (Гастингс Гилфорд). Распространенность синдрома составляет приблизительно 1 на 18 миллионов, таким образом, в любой конкретный момент в мире приблизительно 350-400 детей живут с прогерией.
Диагностика
Прогерия обычно диагностируется в течение второго года жизни или позже, когда прогероидные признаки становятся заметными. Диагноз основывается на тщательной клинической оценке, характерных физических данных, тщательном анамнезе пациента и диагностическом генетическом тестировании.
В более редких случаях заболевание можно заподозрить при рождении на основании распознавания определенных подозрительных данных (например, «склеродермоподобной» кожи над ягодицами, бедрами, нижней частью живота; цианозом средней части лица; «скульптурным» носом).
Для подтверждения или выявления определенных скелетных аномалий, потенциально связанных с расстройством, могут проводиться специализированные тесты. Кроме того, для оценки сопутствующих сердечно-сосудистых нарушений и определения надлежащего ведения заболевания могут также проводиться тщательная оценка состояния сердца и постоянный мониторинг (например, клинические обследования, рентгенологические исследования, специализированные исследования сердца).
Лечение прогерии
В сентябре 2012 года результаты первого в истории клинического исследования лекарственных препаратов для детей с прогерией показали, что Лонафарниб, тип ингибитора фарнезилтрансферазы, первоначально разработанный для лечения рака, оказался эффективным для лечения прогерии. Каждый ребенок продемонстрировал одно из четырех улучшений:
- увеличение веса;
- улучшение слуха;
- улучшение структуры кости;
- повышение гибкости кровеносных сосудов.
Помимо Лонафарниба, который не одобрен еще управлением по санитарному надзору за качеством пищевых продуктов и медикаментов и, следовательно, доступен только для клинических испытаний, лечение синдрома преждевременного старения направлено на конкретные симптомы, проявляющие у каждого человека. Лечение расстройств может потребовать скоординированных усилий группы специалистов, которым может потребоваться систематическое и всестороннее планирование лечения больного ребенка. Такими специалистами могут быть:
- педиатры;
- врачи, диагностирующие и лечащие нарушения скелета, мышц, суставов и других родственных тканей (ортопеды);
- врачи, диагностирующие и лечащие аномалии сердца и кровеносных сосудов (кардиологи);
- физиотерапевты и другие специалисты здравоохранения.
Специальные методы лечения для людей с синдромом являются симптоматическими и поддерживающими. Например, у пациентов с приступами боли в груди из-за недостаточного поступления кислорода в сердечную мышцу (ангинальные приступы) лечение может включать использование определенных лекарств, помогающих минимизировать такие симптомы.
Прогноз
Средняя продолжительность жизни людей с прогерией составляет 14 лет, хотя некоторые люди живут в возрасте старше 20 лет. Прогерия — это смертельный синдром.
Люди с заболеванием подвергаются повышенному риску многих заболеваний (вывихи, переломы, заболевания сердца, инсульт и проч.). У детей с прогерией очень часто развивается атеросклероз и суженные артерий. Большинство больных детей в конечном итоге умирают от болезней сердца.
Источник