
Синдромы наследственной и врожденной патологии

Добрый день! Меня зовут Захарова Ольга Михайловна. Я – врач-генетик, работаю в Центре иммунологии и репродукции.
Сегодня мы поговорим о хромосомных болезнях, о генетических патологиях – какие они бывают и по какой причине появляются?
Какая патология относится к наследственной?
Это патология, связанная с патологией в генах и в хромосомах.
- Первая группа наследственных болезней – это хромосомная болезнь. Хромосомная болезнь связана с аномалиями количества хромосом или их структуры.
Причиной всех наследственных болезней является мутация. Соответственно, к хромосомным болезням приводят хромосомные мутации и так называемые числовые мутации хромосом.
Самые распространенные хромосомные болезни – это те, которые связаны с аномалиями числа хромосом. Всем известный синдром Дауна связан с аномалиями числа хромосом, чаще всего – с лишней 21-ой хромосомой. Здоровые люди имеют 23 пары хромосом, то есть – 46 хромосом. Отличаются мужчины и женщины только по одной хромосоме. Это половая хромосома Y – у мужчин, X – у женщин.
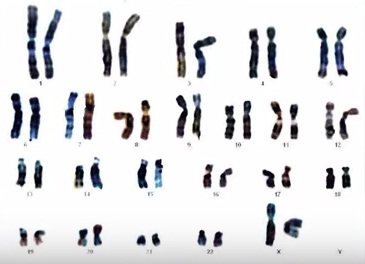
Нормальный кариотип женский записывается как 46,XX ; нормальный хромосомный набор у мужчин – 46,XY . Синдром Дауна, чаще всего, связан с добавочной хромосомой в 21-ой паре, записывается, в зависимости от того, мальчик это или девочка: 47,XX+21; 47,XY+21 . Это указывает на то, что лишняя хромосома находится в 21-ой паре. Мальчики и девочки поражаются в равной степени. И мальчики, и девочки могут иметь аномальный кариотип, соответствующий синдрому Дауна. Синдром Дауна встречается с частотой в среднем 1 на 700 новорожденных.
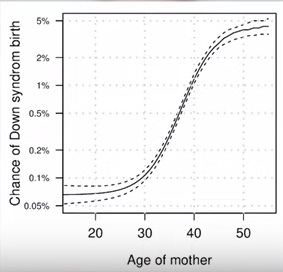
Сейчас всем уже известно, что с возрастом женщины повышается вероятность рождения ребенка с синдромом Дауна. После 36 лет риск переходит так называемую границу порога отсечки, которая при скрининге на графике сплошной горизонтальной линии отделяет низкий риск от высокого риска .
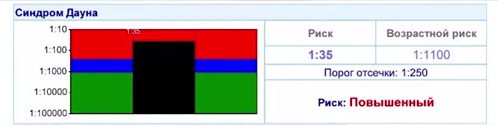
Возраст мужчин критично не влияет на появление ребенка с синдромом Дауна, потому что в процессе оплодотворения участвует большое количество сперматозоидов (миллионы). Если в каких-то сперматозоидах произошла мутация, и они содержат лишнюю хромосому, им труднее конкурировать за яйцеклетку. Как правило, яйцеклетку оплодотворяет самый здоровый, самый хороший, самый генетически достойный сперматозоид.
В процессе оплодотворения чаще всего участвует только одна яйцеклетка. Поэтому, если там произошла мутация – никуда не деться – сперматозоид оплодотворяет эту яйцеклетку. Поэтому, именно возраст женщины является фактором риска рождения ребенка с синдромом Дауна и с другими аналогичными хромосомными синдромами, которые статистически просто встречаются реже, в силу их тяжести.
Потому что синдром Дауна часто, конечно, связан с тяжелыми пороками развития внутренних органов, с тяжелыми аномалиями. Нередко такая беременность заканчивается замиранием, происходит выкидыш или замершая беременность. Но, тем не менее, определенный процент таких беременностей вынашивается, и может родиться ребенок, который впоследствии может даже долго жить. Хотя продолжительность жизни таких людей ограничена в силу разных причин.
- Следующей группой наследственных болезней являются моногенные болезни. Это заболевания, связанные с мутациями в единичном гене.
Таких заболеваний очень много. В настоящее время их описано более 6000. Каталог этих болезней еще не закрыт. Ежегодно происходит описание нескольких таких синдромов.
С другой стороны, происходит объединение многих синдромов в одну группу, потому что выявляется одна и та же мутация. И таким образом многие заболевания объединяют в одну группу. Заболеваний очень много по количеству, но они очень редко встречаются в популяции. Эти заболевания относятся к разряду орфанных или редких заболеваний. В семьях очень часто даже не знают, что это за заболевание и очень удивляются, откуда вдруг оно появилось у ребенка.
Моногенные болезни, орфанные болезни, имеют разные типы наследования: аутосомно-доминантный, аутосомно-рецессивный, сцепленное с полом доминантное и рецессивное.
Доминантное заболевание характеризуется тем, что заболевание передается из поколения в поколение.
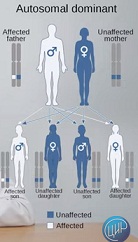
Очевидно, что супруги предполагают наличие у детей такого заболевания, потому что кто-то из них уже имеет клинические проявления данной патологии. Им проще принять решение о рождении ребенка, потому что, если один из родителей имеет это заболевание, если это заболевание не помешало человеку вырасти, получить образование, вступить в брак, то почему им отказываться от деторождения, если ребенок будет такой же, как папа или мама.
Аутосомно-рецессивные заболевания – еще одна группа моногенных или орфанных, которое связано с тем, что каждый из родителей является носителем дефектного гена, но они здоровы. Это парный ген, а все гены у нас парные, точно так же, как и хромосомы, потому что ровно половину генетической информации мы получаем от отца, а половину – от мамы. Родители здоровы, но они имеют в паре генов один дефектный рецессивный ген.
Кстати, все здоровые люди имеют такие дефектные рецессивные гены. Мы о них не знаем, потому что у нас есть здоровые дети, мы сами дети своих здоровых родителей и считаем себя здоровыми. Но то, что мы несем – это надо принимать как аксиому. У родственных супружеских пар вероятность того, что они являются носителями одного и того же дефектного рецессивного гена гораздо выше, чем у супружеских пар не родственных. В половой клетке: в яйцеклетке или в сперматозоиде, примерно 25000 генов. На эти 25000 генов примерно 4-8 генов считаются дефектными рецессивными.
Конечно, это небольшое количество, но у родственных пар вероятность того, что сперматозоид и яйцеклетка будут нести один и тот же дефектный рецессивный ген, конечно, гораздо выше.
Половые клетки несут половинную информацию и в отличие от любой клетки человеческого организма, где 46 хромосом, половые клетки несут только 23 хромосомы, потому что при оплодотворении яйцеклетки сперматозоидом должен восстанавливаться полный генетический состав. То есть 46 хромосом уже несет зигота – оплодотворенная яйцеклетка.
Вернемся к рецессивному наследованию. Аутосомно-рецессивные наследования – это если каждый из супругов имеет один дефектный рецессивный ген в паре. Половые клетки могут образовываться как здоровые, несущие здоровый ген, так и половые клетки, которые несут дефектный рецессивный ген. Отсюда понятно, что может быть здоровая яйцеклетка, которая оплодотворяется сперматозоидом с дефектным рецессивным геном. В данном случае ребенок будет здоров. Он будет нести как здоровый, так и дефектный ген.
Если яйцеклетка будет нести дефектный рецессивный ген, а сперматозоид придет здоровый, тогда, опять же, ребенок будет здоровый. Возможна ситуация, когда оба здоровых гена попадают в зиготу, в оплодотворенную яйцеклетку. То есть и от мамы, и от папы приходит здоровый ген – ребенок будет вообще лишен этого дефектного рецессивного гена.
И последняя ситуация, когда и яйцеклетка, и сперматозоид несут дефектные рецессивные гены. В данном случае нет противовеса в виде здорового гена, и ребенок лишен этой защиты. В данном случае будет развиваться аутосомно-рецессивная патология.
Ситуация, конечно, очень тяжелая, потому что супруги сами здоровы, у них могут быть все родственники здоровы – бабушки, дедушки, тети, дяди, племянники. И вдруг рождается такой ребенок. Мало того, что ребенок будет болен (а нередко аутосомно-рецессивные болезни очень тяжелые), супруги узнают, что это наследственная болезнь. Им трудно это понять: как они, будучи здоровыми сами, имеют ребенка с наследственной патологией. Но и это еще не всё. Оказывается, что вероятность того, что последующие дети будут иметь ту же самую патологию также высока, и относится к высокому генетическому риску. Чисто теоретически вероятность эта равна 25%. В данном случае необходима генетическая диагностика, ДНК-диагностика, молекулярно-генетическая диагностика.
Не все моногенные болезни можно диагностировать такими молекулярно-генетическими методами. Тем не менее, необходимо попробовать провести такую диагностику, и если будет выявлена мутация в гене, тогда возможно проведение пренатальной диагностики на ранних этапах следующей беременности с целью выявления этой патологии. В некоторых случаях возможно лечение. Лечение, чаще всего, симптоматическое, то есть это просто помощь ребенку развиваться, чтобы не было тяжелых последствий.
При некоторых болезнях можно проводить лечение патогенетическое, влияющее на патологические звенья клинической картины, и возможно, если не излечение, то во всяком случае вполне комфортное существование такого ребенка.
В родильном доме всех новорожденных детей обследуют на 5 наиболее часто встречающихся моногенных аутосомно-рецессивных болезней. Именно в родильном доме у ребенка берут кровь из пяточки на тестовые полоски-системы, и уже после выписки из роддома, если что-то выявляют, какую-то патологию (одну из пяти нозологических форм), сообщают родителям и начинают немедленно лечить.

В этом случае, повторяю, ребенок может быть вполне здоров, и у него не будет тяжелых клинических проявлений. Поэтому очень важным является рождение ребенка в родильном доме, потому что домашние роды могут пропустить такой важный аспект, как диагностика, скрининговое обследование ребенка на наследственные генетические болезни.
В настоящее время, конечно, есть метод диагностики гетерозиготного носительства. То есть носительства супружеской парой наиболее распространенных дефектных рецессивных генов. Тем более, если семье уже известно о наличие какого-то из этих заболевания, то можно пройти и такое обследование.
- И, наконец, еще одна группа наследственных заболеваний – это так называемые болезни с наследственным предрасположением. Они по-другому называются многофакторными болезнями, или мультифакториальными, или полигенными болезнями.
Это самая распространенная группа наследственных болезней. Вот эти болезни, как раз, встречаются в каждой семье, эти заболевания есть у каждого человека. Даже если он здоров, то они в перспективе могут быть. Чаще всего, эти заболевания – так называемые возрастные: гипертоническая болезнь, ишемическая болезнь сердца. К таким заболеваниям относится варикозная болезнь вен нижних конечностей. Это заболевания, которые могут поражать любые органы системы: это и гастрит, и язвенная болезнь желудка, двенадцатиперстной кишки. Это желчнокаменная болезнь, это почечнокаменная болезнь, это практически все заболевания психической сферы: шизофрения, эпилепсия. Плоскостопие, сколиоз, даже аппендицит считается заболеванием с наследственным предрасположением, несмотря на то, что это воспаление. Есть семьи, где из поколения в поколение, чуть ли не в одном и том же возрасте, члены семьи попадают на операционный стол с острым аппендицитом.
К группе мультимногофакторной патологии относятся также врожденные пороки развития (изолированные врожденные пороки развития), то есть не связанные с какими-то другими пороками. Например, при хромосомных нарушениях, когда множественные врожденные пороки поражают все органы системы или многие: зрение, центральная нервная система, это желудочно-кишечный тракт, половая сфера, патология скелета и так далее.
Многофакторные пороки – это изолированные. Они тоже встречаются часто. Например, расщелина губы и неба у ребенка или врожденный порок сердца, или врожденный вывих или подвывих тазобедренного сустава, или врожденная косолапость. Также гипоспадия, эписпадия.
Вывих тазобедренного сустава – это врождённая патология, чаще всего поражающая девочек. Врожденная косолапость, наоборот, чаще поражает мальчиков. В данном случае, конечно, желательно обратиться к генетику, потому что, если поражается менее поражаемый пол, например, врожденная косолапость у девочки, то риск для последующих детей в семье будет выше, нежели если врожденной косолапостью страдает первый ребенок мужского пола. Этот риск повышается не только для братьев и сестер, но также и для потомства этого человека. Если девочка с врожденной косолапостью – риск для её детей будет выше, чем если врожденной косолапостью страдал мальчик, который уже вырос, женился и собирается иметь детей.
Эти пороки, эти проблемы устранимы. Но иногда они доставляют очень большие хлопоты и страдания супружеской паре, чаще всего, конечно, маме. Поэтому пренатальная диагностика, которая выявляет многие пороки еще на уровне внутриутробного развития ребенка, помогает родителям, в частности маме, адаптироваться к тем проблемам, которые будут после рождения ребенка.
Это не значит, что родители будут принимать решение о прерывании беременности, нет. Но они будут готовы психологически решать их уже с первых дней рождения ребенка. Это не будет острой психотравмой, потому что можно уже найти информацию в Интернете: где лечить, как лечить, к каким специалистам обращаться, как помочь ребенку. Это тоже очень важно.
.
Это основные группы наследственных болезней, о которых я хотела вам сказать. Есть еще заболевания, тоже относящиеся к наследственным. Благодаря успехам молекулярной генетики стало возможным выделение этих групп в отдельную группу наследственных болезней. Они встречаются гораздо более редко, но тоже имеют свои закономерности. Об этом можно поговорить в следующий раз.
Преимуществом наших клиник является наличие собственной
лаборатории, где Вы можете сдать все необходимые анализы до и во время беременности
Источник
Аномалии сосудов. Наследственные заболевания соединительной ткани – синдром MARFANОдни генетически детерминированные нарушения поражают преимущественно артерии (некоторые касаются только аорты), другие поражают вены и лимфатические сосуды. При некоторых аномалиях нарушается развитие нескольких типов сосудов. Заболевания соединительной ткани (дисплазии) подразделяют на две большие группы: (1) болезни, вызываемые мутациями в одном гене, которые детерминируют повреждение или каким-то образом повреждают компоненты внеклеточного матрикса; (2) болезни, вызываемые внешними факторами, поражающими внеклеточный матрикс, например ревматоидный артрит и системная красная волчанка. К первой группе относят и многие заболевания с поражением ССС. Подверженность к так называемым приобретенным заболеваниям соединительной ткани отчасти определяется генетически. Наследственная патология внеклеточного матрикса. Эти заболевания (< 10 видов) впервые были описаны около 50 лет назад. В настоящее время их число насчитывает 200 различных фенотипов. Многие обзоры и учебники посвящены характеристике фенотииических проявлений, генетических основ и причин таких заболеваний. Некоторые из них служат причиной развития различных артериопатий. Синдром Williams — наследственное заболевание, вызываемое делецией генов в хромосоме 7q 11.23, включая и тот, который кодирует тропоэластии (ELN), описан далее. Наследуемый по аутосомно-доминантному типу надклаианный стеноз аорты, который проявляется такой же артериопатией, как и синдром Williams, ассоциирован с мутациями, затрагивающими только ELN.
При некоторых заболеваниях соединительной ткани кажется, что патология сосудов связана с ослаблением их стенки, например ломкость артерий, наблюдаемая при сосудистой форме синдрома Ehlers-Danlos, вызываемого мутациями гена, кодирующего проколлаген типа III, COI.3A1. Похожие патогенетические механизмы вызывают, вероятно, развитие атипичных случаев аневризмы аорты или пролапса клапана, наблюдаемых у больных с классической формой синдрома Ehlers-Danlos и несовершенным остеогенезом. В то же время нарушения ССС, наблюдаемые при синдроме Marfan, вызванном мутациями гена, кодирующего фибриллин-1 (гликопротеин внеклеточного матрикса), не связаны со «слабостью» соединительной ткани. На оборот, большинство фенотииических особенностей обусловлено нарушениями регуляции активности трансформирующего фактора роста бета (ТСЕ-бета), которая обычно контролируется при связывании с фибриллином. Другие наследуемые по законам Менделя артериопатий, как было недавно показано, вызваны дефектами рецепторов TGF-P. Синдром MARFANЭто аутосомно-доминантное заболевание встречается довольно часто (2-3 случая на 10 тыс. чел.) среди представителей всех рас и этнических групп. Несмотря на установление генетических и биохимических основ заболевания, диагностика синдрома Marfan вне семей с классическими фенотипическими проявлениями базируется в основном на анализе клинических симптомов. Современные критерии разработаны с учетом пораженных органов и систем: органов зрения, скелета, сердца и аорты, других систем организма, а также семейного анамнеза. Наличие более специфичных для синдрома Marfan проявлений, таких как дилатация аорты, расслоение аорты у молодых людей без АГ, эктопия хрусталика и эктазия твердой мозговой оболочки, очевидно, более важно с точки зрения диагностики, чем нарушения, характерные для других заболеваний соединительной ткани и встречающиеся в популяции в целом, например сколиоз, гипермобильность суставов, миопия и пролапс митрального клапана (ПМК). К наиболее характерным нарушениям со стороны ССЗ относятся ПМК и дилатация синусов Valsalva. Клинические проявления этих нарушений в виде митральной peгургитации, аортальной регургитации и расслоения аорты и, при отсутствии лечения, становятся причиной большинства случаев ранней смерти, в результате средний возраст таких пациентов < 40-50 лет. У детей наблюдается тенденция к более тяжелым поражениям митрального клапана, тогда как поражения аорты прогрессируют и наиболее вероятны в подростковом и более старшем возрастах. – Также рекомендуем “Наследственное поражение митрального клапана. Наследственное поражение корня аорты” Оглавление темы “Наследственные заболевания сердечно-сосудистой системы”: |
Источник
Семейная акрогерия Готтрона (Acrogeria familiaris Gottron)
Эта акромикрия, сопровождающаяся атрофией кожи конечностей, является наследственной и семейной. Кожа верхних конечностей, особенно кистей, истончается, сморщивается и приобретает желтовато
беловатую окраску со слегка розоватым оттенком. Сквозь атрофическую кожу просвечивают сосуды.
Такие изменения присущи прогерии (Gilford), но при последней общая дистрофия и кожные атрофии генерализованы (ранняя старость), и она не носит семейного характера.
Gerodermia infantum (geromorphismus cutis; progeria)
Это синдром врожденных дистрофий, проявляющихся сморщенной, истонченной кожей, гипоплазией половых органов, евнухоидизмом, замедлением роста и развития интеллекта, пресенильным нанизмом мышечной атрофией, гиперпигментацией и изменениями костей.
К этим дистрофиям можно отнести синдром HutschinsonGilfordа (progeria со старческим нанизмом), акрогерию и некоторые проявления ранней старости.
«Синтетическая дерматология»,
Любен Попов
Onycho-arthro-osteodysplazia hereditaria; syndroma Оesterreicher Заболевание представляет собой наследственную и семейную полидисплазию, характеризующуюся дистрофией ногтей или анонихией, наиболее выраженной на больших пальцах и аномалиями суставов и костей — аплазией надколенных чашек, уплощением наружных мыщелков большеберцовых костей, искривлением малоберцовых костей, дисплазией лучевых костей с вывихом, гипоплазией плечевых костей, ключиц, костей таза, позвоночника и др. Это сложная дисплазия передается…

Представляет собой трофоневроз, проявляющийся гемиатрофией лица у детей и юношей. Начинается с появления синевато-темных пигментированных пятен, а иногда с уменьшения пигментации кожи на одной половине лица (главным образом в области глаза, щеки, нижней челюсти). Затем развивается прогрессирующая склероатрофия кожи и атрофия жевательных и глазных мышц и языка с дистрофией и выпадением зубов и ресниц. Часто…
Этот редкий, доминантно передающийся синдром проявляется ре-цидивирующими буллезными высыпаниями на стопах и кистях, сходными с буллезным эпидермолизом. Рецидивы наступают спонтанно, в особенности после действия холода или тепла. Как правило, заболевание начинается еще в детстве, но наблюдались случаи более позднего его развития. Повидимому, патогенетически оно связано с наследственным предрасположением и присоединяющимися внешними травматическими факторами. Таким образом,…

Описанный WerneroM в 1904 году этот синдром проявляется катарактой (развивается к 20 — 25 годам); развитием диффузной склеродермии или склеропойкилодермии типа Arndt-Jaffe, начинающихся после периода полового созревания (в 16 — 30 лет), причем кожный процесс прогрессирует, поражаются конечности и лицо; на нижних конечностях могут развиться язвы и явления гиперкератоза; дистрофией ногтей и ранним поседением; охриплостью,…
Этот синдром состоит в сочетании врожденного порока сердца, цианоза губ и ложа ногтей, искривления пальцев, телеангиэктазий на щеках и покраснения десен. Явления со стороны кожи и слизистых оболочек весьма важны с диагностической точки зрения — при обследовании выявляется врожденный порок сердца; излечение наступает после хирургического устранения дефектов сердца.Гкомедоновый невусNaevus comedonicus, naevus acneiformis unilateralis; Syndroma cataracto-comedonicum…
Плиткообразный невус, эластический невус Левандовского Соединительнотканные невусы чрезвычайно редкое явление. Они локализуются на всех областях тела, чаще на конечностях и грудной клетке. Иногда они располагаются систематизировано, полосками. Представляют собой небольшие лихеноидные или полусферические плотные папулы, желтовато-белого или коричневого цвета, образующие множественные, обычно наслаивающиеся друг на друга элементы, располагающиеся группами, в виде бляшек с неправильными очертаниями,…
lichen variegatus; parapsoriasis lichenoides Дерматоз начинается у детей старше трех лет незначительными эритематозными и кераготическими изменениями на коже конечностей, туловища и отчасти лица, сопровождается образованием фолликулярных роговых папул, сухостью кожи и волос и изменениями ногтей. В стадии полного развития обнаруживают выступающие над уровнем окружающей кожи тесно, линейно расположенные роговые конусовидные узелки, как бы погруженные в…
Представляет собой добавочные, неразвитые, зачаточные молочные железы, располагающиеся линейно по направлению от молочных желез вниз. Это проявление атавизма может быть в виде единичной или нескольких желез. Иногда такая недоразвитая железа обнаруживается на бедре или в области ягодицы.Порокератоз Мибелли (Porokeratosis Mibelli) Типичная форма этого заболевания выражается появлением округлых полициклических сухих кератотических очагов поражения, рассеянных или одиночных, иногда…
Syndroma osteo-cutaneo-endocrinopathica; syndroma albright Этот полисимптомный врожденный синдром развивается в раннем детском возрасте или к периоду половой зрелости. На коже появляются множественные пигментные пятна коричневого или темного цвета, неправильной формы, различной величины; они локализуется в области груди или верхних конечностей, а иногда и в других местах. Отмечаются опухоли и деформации костей черепа, позвоночника и таза.…
Syndroma ataxoteleancilf ctatlcum Болезнь начинается в раннем детском возрасте .и достигает своего развития к 5 — 6 году жизни. Вначале нарушения двигательной функции выражены неустойчивым стоянием и походкой, трудно сохраняемым равновесием, несогласованностью волевых движений, дисимметрией и паданием при попытке активных движений. В спокойном состоянии у таких детей наблюдаются непроизвольные хореиформные и дистрофические движения. Затем начинают постепенно…
Источник