
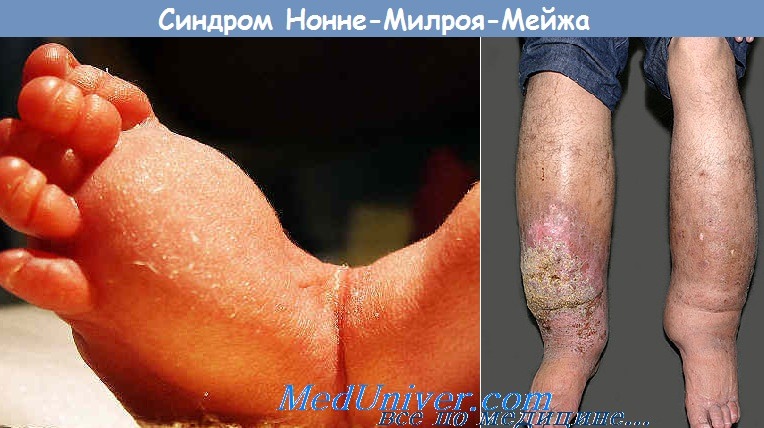
Синдромы нонне милроя и мейжа

Синдром Нонне-Милроя-Мейжа (Nonne-Milroy-Meige) – синонимы, авторы, клиникаСинонимы синдрома Нонне-Милроя-Мейжа. S. (M.) Meige. S. Meige— Milroy. S. (M.) Nonne—Milroy. M. Milroy. Хроническая наследственная трофодерма. Хроническая трофодерма. Врожденная трофодерма. Лимфатический отек (Virchow). Псевдоотечная гиподермальная гипертрофия. Врожденная наследственная слоновость (Nonne). Наследственный отек. Идиопатический наследственный лимфатический отек. Трофоневроз. Невроартритическая псевдослоновость. Семейный наследственный отек. Определение синдрома Нонне-Милроя-Мейжа. Семейный хронический трофический отек (слоновость). Авторы. Meige Henry — французский врач, Париж, 1866—1940. Milroy William Forsyth — американский терапевт, Охама (штат Небраска), 1855 —1942. Nonne Мах — немецкий невропатолог, Гамбург, 1861—1959. Впервые синдром описал Virchow в 1863 г. под названием «лимфатический отек с пороками развития». Meige описал симптоматику в 1889 г., Nonne — в 1891 г. и Milroy — в 1892 г. Никто из этих трех авторов, однако, не установил в последующих описаниях связи симметричного трофического отека с эндокринными и другими общими расстройствами. Симптоматология синдрома Нонне-Милроя-Мейжа: Этиология и патогенез синдрома Нонне-Милроя-Мейжа. Страдание часто носит семейный характер. По-видимому, речь идет о развивающихся в эмбриональном периоде генетических (нерегулярное доминантное наследование, иногда сцепленное с полом) или перистатических нарушениях системы гипофиз — промежуточный мозг. Связь с S. Laurence— Moon—Biedl—Bardet (?). Дифференциальный диагноз. S. Laurence — Moon—Biedl—Bardet (см.). S. Frohlich (см.). Лимфангиэктатический отек. S. Bonnevie—Ullrich (см.). S. Ullrich—Turner (см.). Слоновость другого происхождения.
– Также рекомендуем “Синдром Нормана-Вуда (Norman-Wood) – синонимы, авторы, клиника” Оглавление темы “Синдромы в медицине”:
|
Источник
- Врачам /
- Полезные статьи /
- Пренатальная ультразвуковая диагностика болезни Нонне-Милроя
Д.В. Воронин, И.В. Чубкин.
Врожденная (наследственная) лимфедема I типа (болезнь Нонне-Милроя)- достаточно редкое аутосомно-доминантное
заболевание с неполнойпенетрантностью [Makhoul IR, Козлова С.И.].
Первые публикации о БНМ,принадлежат М. Nonne (немецкий невропатолог 1861-1959гг.), и в 1892 г. -американскому
врачу W.F. Milroy (1855-1942 гг.) [Milroy W.F.].
Причиной заболевания является мутация внутри каталитической петликиназной области VEGFR-3 (первого молекулярно
идентифицированногомаркера, специфически экспрессирующегося в лимфатическом эндотелии),вызывающая снижение
аутофосфорилирования рецептора лимфатическогоэндотелия, ингибирующего активность рецепторов и уменьшающегопередачу
сигналов пролиферации и роста лимфатических сосудов. Makhoul I.R. и соавт. (2006), обобщая данные о причинах
БНМ,указывают на повреждения в локусе VEGFR-3, приводящего в конечномитоге к дисгенезии лимфатических сосудов с
нарушением лимфооттока отнижних конечностей, причем степень повреждения микроциркуляторногорусла может начинаться
ассиметрично, с одной ноги [Makhoul I.R]. SukolinG.I. и PasportnikovaO.A. (1990)связывают возникновение CНМ с
приемом больших доз сульфаниламидов нафоне беременности (описан случай лечения бронхита в первом
триместребеременности, вызвавший спорадический случай возникновения СНМ уплода).
Клинически БНМ характеризуется
наличием хронического отеканижних конечностей (как фрагмента, так и всей ноги).
Эхографически БНМ проявляется локальным увеличением подкожнойскладки (подкожной клетчатки), представленной
достаточно гомогеннойзоной пониженной эхоплотности на уровне голеней и стоп плода. БНМможет протекать в сочетании с
агенезией мозолистого тела игипертелоризмом. Некоторые авторы небезосновательноприводят к общему знаменателю такие
ультразвуковые признаки, какувеличение толщины воротникового пространства и лимфедему нижнихконечностей,
диагностированную уж е в I триместре беременности.
МАТЕРИАЛЫ И МЕТОДЫ
Беременная З., 31 год, была направлена в МГЦ с целью проведенияочередного скринингового УЗИ,
по данным первичной скринирующейэхографии, выполненной в женской консультации, патологии выявлено небыло.
Наследственность беременной отягощена семейной врожденнойлимфедемой I типа (БНМ), клинические проявления которой
(отечностьнижних конечностей) пенетрировали у нескольких поколений родственниковбеременной.
Ультразвуковое исследование проведено на приборесиспользованием трансдьюссеров: трансабдоминального 3,5-5,5 МГц
илинейного 5,0-7,5 МГц.
РЕЗУЛЬТАТЫ
В процессе ультразвукового исследования выявлен один живой плодмужского пола, соответствующий сроку
33 недели беременности и имеющийэхографические признаки двухстороннего диффузного утолщения мягкихтканей голеней и
стоп до 11 мм. Наосновании эхографических данных у плода и учитывая семейный анамнезбеременной, был заподозрен
врожденный наследуемый порок развитиялимфатической системы: двухсторонний лимфостаз области голеней и стоп.После
проведения консилиума в составе врача УЗД, генетика и ангиолога(хирурга-флеболога), учитывая благоприятный прогноз
БНМ (лимфедема Iтипа), беременность была пролонгирована.
Беременность завершилась естественными родами на доношенномсроке. Родился живой, доношенный мальчик массой 3400
гр., длиной 51 см.,с оценкой по шкале Апгар 8/9 баллов. После родов пренатальнопоставленный диагноз подтвержден,
при этом выявлены: двухсторонняяотечность голеней и стоп новорожденного, сохранившиеся в нео- ипостнатальном
периодах онтогенетического развития данного пациента.Ребенок проконсультирован врачом-генетиком, поставлен диагноз:
«БолезньНонне-Милроя (врожденная лимфедема I типа)».
ОБСУЖДЕНИЕ
Известно, что не всякий отек нижних конечностей являетсяпроявлением БНМ, что предусматривает необходимость
построениядифференциально-диагностического ряда при обнаружении симптомов БНМ.
Отличительным признаком БНМ является наличиеотеков только нижних конечностей, а при лимфедеме II типа (болезнь
Нонне-Милроя-Мейджа) могут появляться отеки верхних конечностей, лица игрудной клетки, что может быть подтверждено
уменьшением числа не толькопаховых, но и подмышечных лимфоузлов при диагностической лимфографии. Этот факт
представляется крайне важным, потому чтопрогноз для жизни при лимфедеме I типа – благоприятный, а при лимфедеме II
типа может быть сомнительным.
Прогноз для истинного БНМ – благоприятный.Однако осложнения, связанные с
хроническим отеком нижних конечностей,могут существенно изменить качество жизни пациента. Характерныеосложнения
при БНМ – это нарушение местной терморегуляции (повышениетемпературы области лимфедемы), локальное
изменениесветочувствительности (возможно возникновение тяжелых ожогов, в томчисле вследствие солнечного загара),
васкулиты, целлюлиты, инфекционныезаболевания кожи и расположенных под ней тканей (в том числе erysipelas), атак
же последствия пункций лимфоузлов.
Прогресс перинатальной медицины невозможен без решения основныхвопросов метаболизма в системе
«мать-плацента-плод». Поэтому синдромальный подход ицеленаправленный поиск родительских аномалий у плода является
важнойособенностью проведения пренатальнойэхографии при наличии в семье наследуемых заболеваний. С точки
зренияпрактической значимости метода УЗД, наш случай еще раз подтверждаетэтот тезис. Поэтому мы считаем важным
предусматривать диагностическуюнеобходимость и техническую возможность врача УЗД расширятьстандартный объем
пренатальнойэхографии (фетометрии плода) оценкойнеобходимых (диагностически и клинически значимых) структур
(органов –тканей – морфологических единиц) организма пробанда. Это позволит ближеподойти к адекватной оценке причин
выявленных изменений иформированию правильного прогноза для будущего потомства.
© Клиника плода и мамы доктора Чубкина, 2012-2018
ООО «УК
«АРНИКА», ОГРН 1167847280281, ИНН 7805677133
Лицензия № ЛО-78-01-008480 от 10 января 2018 года
Информация на сайте не является публичной офертой и носит справочный
характер. Имеются противопоказания.
Источник
Max Nonne (1861-1959)
Wiliam Forsyth Milroy (1855-1942)
HenriMeige (1866-1940)
Син.: наследственная лимфедема.
Наследственное заболевание, характеризующееся развитием трофедемы лица и/или конечностей. В настоящее время принята классификация, согласно которой выделяют два типа наследственной лимфедемы: тип I (болезнь (Нонне–)Милроя, или первичная врождённая лимфедема) и тип II (болезнь Мейжа, или поздняя лимфедема).
Наследственная лимфедема I типа обусловлена мутацией гена FLT4 (локус 5q35.3), кодирующего рецептор фактора роста эндотелия сосудов-3. Большинство случаев характеризуется аутосомно-доминантным типом наследования с неполной пенетрантностью (около 84%), вариабельной экспрессивностью и возрастом дебюта (от рождения до 35 лет). Заболеваемость – 1:6000 новорождённых; соотношение полов 1:2,3. Как правило, дебют заболевания – с рождения или сразу же после рождения. У ребёнка отмечается дистальный плотный тёплый отёк конечностей, безболезненный, без тенденции к изъязвлению и варикозу. В большинстве случаев отёк сохраняется на всю оставшуюся жизнь; изредка возможно спонтанное выздоровление.

Трофедема верхней конечности у новорождённого с болезнью Нонне — Милроя (источник: https://www.thefetus.net/images/case-images/case0066_files/case0066-8.jpg)

Трофедема нижней конечности у новорождённого с болезнью Нонне — Милроя (источник: https://www.thefetus.net/images/case-images/case0066_files/case0066-10.jpg)

A
B
Пациенты с болезнью Нонне – Милроя: девочки 5 (рис.А) и 7 (рис.B) лет (источник: А – https://www.aik-info.de/zzz_history_kl/Elephantiasis/Maedchen5.jpg; В — https://www.aik-info.de/zzz_history_kl/Elephantiasis/Maedchen7.jpg)
Возможно также развитие хилёзного асцита, гидроторакса, хотя в целом для заболевания не характерны сопутствующие аномалии. Прогноз обычно благоприятный, заболевание в основном характеризуется косметическим дефектом. Описаны сочетания лимфедемы с гидроцеле, дефектами межпредсердной перегородки, аномалиями развития лицевого черепа. Из осложнений описаны лимфангиэктазия кишечника, рецидивирующий септический артирит, (лимф)ангиосаркома.
Впервые наследственная лимфедема нижних конечностей была описана немецким неврологом Максом Нонне в 1891г. (Nonne M. Vier Fälle von Elephantiasis congenita hereditaria // [Virchows] Archiv für pathologische Anatomie und Physiologie und für klinische Medicin, Berlin, 1891. – S.189-196). Американский невролог Уильям Милрой в 1892г. описал 22 случая заболевания (Milroy W.F. An undescribed variety of herditary edema // New York Medical Journal, 1892. – Vol.56. – P.505-508). Эпонимическое название «болезнь Милроя» было предложено английским врачом сэром Уильямом Ослером в его учебнике «The Principles and Practice of Medicine», изданном в 1892г. в Нью-Йорке. Термин «болезнь Нонне – Милроя» получил распространение в немецкоязычной литературе.
Наследственная лимфедема II типа обусловлена мутацией гена FOXC2 (локус 16q24.3), кодирующего один из факторов транскрипции. К аллельным заболеваниям лимфедемы Мейжа относятся сочетание лимфедемы с дистихиазом (двойной ряд ресниц), птозом, жёлтыми ногтями. Наследуется по аутосомно-доминантному типу, дебют после полового созревания. Описаны сочетания поздней лимфедемы с нейросенсорной глухотой, первичной лёгочной гипертензией, мальформациями сосудов головного мозга, экстрадуральными кистами, аномалиями развития позвоночника. Клинически характеризуется развитием трофедемы самой разнообразной локализации, более выраженной ниже пояса.

Схематическое изображение различных локализаций трофедемы при болезни Мейжа (страница из классической работы Анри Мейжа 1899г.) (источник: https://www.aik-info.de/zzz_history_kl/Elephantiasis/meige1899a.jpg)

Пациентки с болезнью Мейжа, 17 и 21 лет (страница из классической работы Анри Мейжа 1899г.) (источник: https://www.aik-info.de/zzz_history_kl/Elephantiasis/Meige1899.jpg)
Довольно часто пациентов беспокоят выраженные боли. Возможно вовлечение в процесс тканей лица и гортани. Несколько выше, чем в популяции, у таких больных риск развития злокачественных новообразований.
Заболевание впервые описано Рудольфом Вирховым в 1863г. В работах французского врача Анри Мейжа была подробно описана клиника болезни и указан её наследственный характер (MeigeH. Dystrophieoedematosehereditaire // Lapressemédicale, Paris, 1898. – Vol.6. – P.341-343) (MeigeH. Letrophoedèmechroniquehéréditaire // NouvelleiconographiedelaSalpêtrière, Paris, 1899. – Vol.12. – P.453-480).
Источник

Наследственная лимфедема – это группа генетических патологий, которые проявляются нарушением лимфооттока от различных участков тела (чаще всего – нижних конечностей) с развитием выраженного отека. Симптомами этого состояния является отечность пораженных конечностей, развивающаяся в различном возрасте (в зависимости от формы заболевания), усугубляющаяся при физической нагрузке и под воздействием иных факторов. Диагностика наследственной лимфедемы производится на основании данных наследственного анамнеза пациента и физикального осмотра. В отношении некоторых типов патологии разработаны молекулярно-генетические методы определения. Специфического лечения не существует, для ослабления выраженности симптомов используют различные техники лимфодренажного массажа и специальные диеты. В некоторых случаях проводят хирургическую коррекцию.
Общие сведения
Наследственная лимфедема (первичная лимфедема, слоновость) – группа врожденных нарушений лимфообращения, которые могут быть обусловлены дисплазией лимфатических сосудов, аномалиями процесса резорбции тканевой жидкости или разрушением клапанов. Это заболевание известно с глубокой древности, но его подробное изучение началось только в XIX веке – например, в 1891 году У. Милрой описал семейную форму наследственной лимфедемы, (годом ранее подобную патологию описал Нонне, но не смог доказать ее врожденную природу). В последующие годы было открыто множество форм этого состояния с различной степенью выраженности симптомов. Были описаны как изолированные типы, проявляющиеся только отеком, так и варианты патологии, сочетающиеся с другими нарушениями.
Наследственная лимфедема встречается достаточно часто относительно других наследственных патологий, однако ее точная распространенность не определена, установлено лишь, что для некоторых форм эти цифры составляют порядка 1:10000. Среди больных преобладают женщины, по данным специалистов их доля в общем количестве пациентов с отеками генетической природы составляет около 80%. Чаще всего наследственная лимфедема проявляется в детском и подростковом возрасте, поэтому большинство больных – молодые люди младше 18-ти лет, однако некоторые формы патологии характеризуются более поздним развитием.

Наследственная лимфедема
Причины
Наследственная лимфедема любого типа возникает по причине нарушения функционирования лимфатической системы – это может быть дисплазия лимфатических сосудов, разрушение их клапанов или нарушение процессов реабсорбции тканевой жидкости. Большинство форм этого состояния является аутосомно-доминантными наследственными заболеваниями, в ряде случаев наблюдается неполная пенетрантность дефектного гена, особенно у гетерозигот. Превалирующее количество женщин в половом распределении наследственной лимфедемы обусловлено физиологическими и эндокринными особенностями – наличие повышенного уровня эстрогена снижает давление тканевой жидкости, что облегчает ее накопление. К такому состоянию могут приводить мутации различных генов, что также отражается на фенотипической картине заболевания.
Классификация
Выделяют следующие наиболее распространенные формы наследственной лимфедемы:
- Синдром Нонне-Милроя или наследственная лимфедема типа 1А – в отношении причин этого заболевания с аутосомно-доминантным типом наследования врачи-генетики еще не пришли к однозначному выводу. Выяснено, что дефектный ген располагается на 5-й хромосоме, предположительно это VEGF3 – он кодирует один из факторов роста лимфатических сосудов и массово синтезируется в период эмбрионального развития, а у взрослых людей обнаруживается в лимфатическом эндотелии. Дефект этого белка приводит к наследственной лимфедеме по причине дисплазии и недоразвития лимфатических сосудов малого калибра.
- Наследственная лимфедема типа 1В – наименее изученная форма заболевания, которую связывают с мутациями генов, расположенных на некоторых локусах 6-й хромосомы.
- Наследственная лимфедема типа 1С – самый распространенный вариант данной патологии, по некоторым данным он обуславливает более 90-94% всех случаев этого отечного состояния. Причиной развития становятся мутации гена GJC2, локализованного на 1-й хромосоме. Продуктом его экспрессии является белок коннектин-47, представитель широкого семейства протеинов, формирующих щелевые контакты между клетками различных тканей. Коннектин-47 выполняет эту роль в эндотелии лимфатических сосудов, поэтому при дефектах гена GJC2 его проницаемость изменяется, и нормальный отток тканевой жидкости становится затрудненным, что приводит к наследственной лимфедеме. По некоторым данным, при мутации вышеуказанного гена имеет место гипоплазия лимфатических сосудов, но специалисты полагают, что это вторичное явление. Как и многие другие формы этого заболевания, наследственная лимфедема типа 1С передается по аутосомно-доминантному механизму с неполной пенетрантностью.
- Синдром Мейджа или наследственная лимфедема типа 2 – достаточно редкая форма заболевания, характеризующаяся поздним началом, иногда встречаются и детские варианты. Это дает некоторым исследователям повод разделять синдром Мейджа на два подтипа – с ранним и поздним развитием. Этиология данного варианта наследственной лимфедемы досконально не изучена, предполагается мутация гена F0XC2, являющегося одним из факторов транскрипции других генов. В результате нарушается функционирование клапанов лимфатических сосудов, что задерживает транспорт жидкости и создает условия для ее накопления в тканях.
Кроме того, специалисты нередко выделяют в отдельные виды наследственной лимфедемы отеки лимфатической природы, сопровождающие некоторые другие генетические патологии. Наиболее известным вариантом такого состояния является лимфедема при болезни Шерешевского-Тернера. Кроме того, лимфатические отеки могут сопровождать такие заболевания, как синдром желтых ногтей и болезнь Хеннекама.
Симптомы наследственной лимфедемы
Различные варианты болезни характеризуются неодинаковым возрастом появления симптомов, их выраженностью и сопутствующими проявлениями. Например, болезнь Нонне-Милроя сопровождается незначительным отеком ног, который можно выявить уже при рождении больного. В дальнейшем это состояние прогрессирует, приводя к характерной клинической картине заболевания.
Наследственные лимфедемы типов 1В и 1С очень схожи между собой и начинают развиваться в детском или подростковом возрасте. В некоторых случаях отеки ног (реже – рук) могут возникать при этих формах и у взрослых людей. Это обусловлено тем, что недостаточность лимфооттока может частично компенсироваться, не привлекая к себе внимания долгие годы и проявляясь лишь незначительной или умеренной отечностью тканей после физических нагрузок. Беременность, ожирение и неправильное питание способны привести к декомпенсации этих форм наследственной лимфедемы. Синдром Мейджа чаще всего возникает в возрасте 30-50-ти лет, иногда ранее – это связано с постепенным разрушением клапанов лимфатических сосудов.
При дальнейшем течении наследственной лимфедемы образуется порочный круг – нарушения лимфооттока приводят к отеку, который еще сильнее затрудняет работу лимфатических сосудов. Сначала конечности сильно отекают, при надавливании на ткани остается след от пальца. Затем на поверхности кожных покровов ног или рук образуется так называемая «лимонная корочка» – признак застоя лимфы в дерме. Постепенно отек и кожные нарушения прогрессируют. Пораженные конечности заметно увеличиваются в объеме, иногда достигая значительных размеров (слоновость), на коже формируются участки гиперкератоза.
Застой лимфы при наследственной лимфедеме затрудняет микроциркуляцию и питание тканей, поэтому на ногах или руках возникают трофические нарушения (язвы, эрозии), которые часто осложняются вторичной инфекцией. Инфекционные агенты способны поражать еще функционирующие лимфатические сосуды (лимфангит), что еще сильнее затрудняет отток лимфы и ведет к быстрому прогрессированию заболевания. В тяжелых случаях и при отсутствии лечения наследственная лимфедема уродует внешний вид конечностей, может приводить к сепсису и сердечно-сосудистой недостаточности.
Диагностика
Определение лимфедемы вообще как отечного синдрома, как правило, не представляет особой сложности – для этого достаточно простого осмотра кожных покровов больного и пальпации пораженных конечностей. При этом определяется выраженный отек, при надавливании на ткани остаются следы в виде ямок, которые долго не расправляются. При расспросе больного наследственной лимфедемой выясняется, что отечность усиливается при длительной ходьбе, стоянии и физической нагрузке. В некоторых случаях подозрение на это состояние может возникнуть у врача-неонатолога при осмотре новорожденного – например, при болезни Нонне-Милроя. Для доказательства лимфатической природы отеков могут производить такое исследование, как лимфангиографию (контрастное рентгенологическое изучение лимфатических сосудов) – при этом состоянии будет выявляться их гипо- или аплазия.
Наличие в наследственном анамнезе пациента подобных отеков и синдромов у близких родственников указывает на генетическую природу данного состояния. Окончательный диагноз может быть подтвержден молекулярно-генетическими исследованиями – в настоящее время в лабораториях производится определение наследственной лимфедемы, обусловленной мутациями генов GJC2 и VEGF3. При этом совершают прямое секвенирование последовательности вышеуказанных генов, что позволяет обнаружить генетический дефект. В некоторых случаях имеет смысл произвести проверку на иные наследственные патологии, которые, помимо прочего, могут проявляться и отечным синдромом.
Лечение наследственной лимфедемы
Терапия наследственной лимфедемы сводится к нескольким принципам – улучшению оттока лимфы, снижению задержки тканевой жидкости и устранению вторичных нарушений, обусловленных отечным синдромом. Для улучшения оттока применяют разнообразные лимфодренажные массажи (как ручные, так и аппаратные) и особый компрессионный трикотаж, иногда используют местные лекарственные средства. Для некоторых форм наследственной лимфедемы могут быть эффективными хирургические методики, создающие более полноценные пути эвакуации тканевой жидкости в лимфатическую или кровеносную системы. Для снижения задержки лимфы в тканях назначают специальные диеты с пониженным содержанием белков и солей – эти нутриенты способны значительно усугублять течение лимфатического отека.
При запущенных формах наследственной лимфедемы с выраженными вторичными нарушениями (слоновостью, трофическими язвами, инфекцией) проводят лечение осложнений. Для борьбы с инфекцией назначают антибиотики, трофические нарушения лечат при помощи витаминотерапии, регенеративных средств и биогенных стимуляторов. При выраженной слоновости иногда производится хирургическое удаление лишнего объема тканей (резекционное оперативное вмешательство), что приводит к улучшению как внешнего вида, так и состояния больного наследственной лимфедемой.
Прогноз и профилактика
Прогноз наследственной лимфедемы зависит от выраженности симптомов заболевания, своевременности начала и характера лечения, а также ряда других факторов. При оптимальных условиях, регулярных лимфодренажных массажах, правильной диете, ограничении потребления жидкости и физической нагрузки прогноз довольно благоприятный – больных могут беспокоить только отеки ног в конце дня. В случае более тяжелого течения наследственной лимфедемы, нарушении плана лечения или отсутствия терапии данное состояние может привести к инвалидизации больного по причине слоновости и даже к летальному исходу, обусловленному вторичной инфекцией и трофическими нарушениями. Поэтому при выявлении заболевания больным следует тщательно придерживаться всех назначений врача, не употреблять соленую или высокобелковую пищу, следить за питьевым режимом и избегать длительной нагрузки на конечности, особенно статического характера.
Источник