
Синдромы при лишней хромосоме х

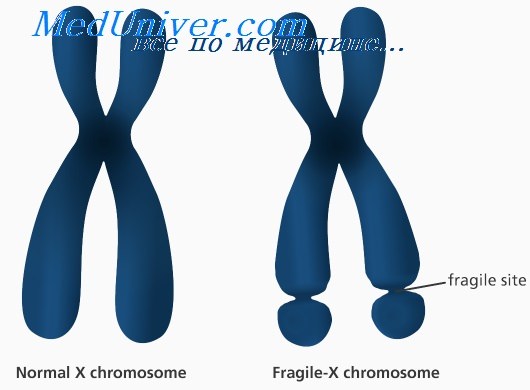
Синдром ломкой Х-хромосомы: причины, диагностика, лечениеЭтиология и встречаемость синдрома ломкой Х-хромосомы. Синдром ломкой Х-хромосомы (MIM №309550) — Х-сцепленное заболевание с задержкой умственного развития, вызванное мутациями в гене FMR1 в Xq27.3. Синдром ломкой Х-хромосомы встречается с частотой 16-25 на 100 000 в общей популяции среди мужчин и в два раза реже среди женщин. Синдром ломкой Х-хромосомы составляет 3-6% всех случаев умственной отсталости среди мальчиков с положительным семейным анамнезом по умственной отсталости при отсутствии врожденных пороков. Патогенез синдрома ломкой Х-хромосомыПродукт гена FMR1, FMRP, экспрессируется во многих типах клеток, но наиболее сильно в нейронах. FMRP может сопровождать определенный подкласс мРНК от ядра к рибосомам. Более 99% мутаций в гене FMR1 — экспансия нуклеотидного повтора (CGG)n в 5′-нетранслируемом участке гена. В нормальных аллелях FMR1 число повторов CGG составляет от 6 до приблизительно 50. В патогенных аллелях (или при полных мутациях) количество повторов более 200. Аллели с более чем 200 повторами CGG обычно имеют гиперметилированную последовательность повторов CGG и смежного промотора FMR1. Гиперметилирование инактивирует промотор FMR1, вызывая снижение экспрессии FMRP. Полные мутации возникают из аллелей премутации (от 59 до 200 повторов CGG) с передачей мутантного аллеля FMR1 от матери (но не от отца); фактически при отцовской передаче премутации часто, наоборот, сокращаются. Полные мутации не могут возникать из нормальных аллелей. Поскольку длина неустойчивых повторов CGG увеличивается в каждом последующем поколении, если они передаются женщиной, обычно наблюдается увеличение числа пораженных потомков в последующих поколениях в семье; этот феномен называется генетической антиципацией. Риск экспансии премутации в полную мутацию возрастает с увеличением числа повторов в премутации. Тем не менее не все премутации одинаково предрасположены к экспансии. Хотя премутации встречаются сравнительно часто, переход в полную мутацию наблюдают только в ограниченном количестве гаплотипов, т.е. когда есть склонность гаплотипа к экспансии. Эта склонность гаплотипа частично может быть связана с присутствием нескольких триплетов AGG, вставленных в последовательность повторов CGG; оказывается, такие триплеты AGG тормозят экспансию повторов CGG, следовательно, их отсутствие в некоторых гаплотипах может предрасполагать к экспансии.
Фенотип и развитие синдрома ломкой Х-хромосомыСиндром ломкой Х-хромосомы вызывает умеренную умственную отсталость у мужчин и легкую умственную задержку у женщин. Наиболее пораженные индивидуумы также имеют поведенческие аномалии, включая гиперактивность, размахивание руками, истерики, плохой зрительный контакт и признаки аутизма. Физические характеристики мужчин изменяются с пубертатом. До полового созревания пораженные мальчики имеют несколько увеличенный размер головы и некоторые другие неотчетливые симптомы; после наступления половой зрелости у них частые более отчетливые признаки (длинное лицо с выдающейся челюстью и лбом, крупные ушные раковины, макроорхидизм). Поскольку эти клинические признаки не уникальны для синдрома ломкой Х-хромосомы, диагноз зависит от молекулярного обнаружения мутаций. Пациенты с синдромом ломкой Х-хромосомы имеют нормальную продолжительность жизни. Почти все мужчины и 40-50% женщин, унаследовавших полную мутацию, будут иметь синдром ломкой Х-хромосомы. Тяжесть фенотипа зависит от мозаицизма метилирования повторов и их числа. Поскольку полные мутации неустойчивы, некоторые пациенты имеют смесь клеток с числом повторов, колеблющимся от премутации до полной мутации (мозаицизм числа повторов). Все мужчины с мозаицизмом числа повторов больны, но часто имеют более высокие показатели умственного развития, чем пациенты с полной мутацией в каждой клетке; у женщин с мозаицизмом числа повторов клинические проявления варьируют от нормы до полного проявления. Аналогично некоторые пациенты имеют смесь клеток с метилированием повторов CGG и без него (мозаицизм метилирования повторов). Все мужчины с мозаицизмом метилирования больны, но часто имеют более высокие показатели умственного развития, чем с гиперметилированием в каждой клетке; женщины с мозаицизмом метилирования также могут быть здоровыми или больными.
Очень редко пациенты имеют полную мутацию, неметилированную во всех клетках; независимо от пола, степень тяжести у них варьирует от нормы до полной клиники. Кроме того, у женщин фенотип зависит от степени смещения инактивации Х-хромосомы. Носительницы премутации (но не полных мутаций) имеют 20% риск ранней дисфункции яичников. Мужчины-носители премутации имеют риск развития синдрома FXTAS. FXTAS проявляет себя как поздняя прогрессирующая мозжечковая атаксия с интенционным тремором. У больных могут также присутствовать снижение краткосрочной памяти и двигательных функций, когнитивные нарушения, а также паркинсонизм, периферическая нейропатия, проксимальная мышечная слабость нижних конечности и дизавтономия. Пенетрантность FXTAS зависит от возраста, обнаруживается в 17% в течение шестого десятилетия жизни, в 38% в течение седьмого десятилетия, в 47% в течение восьмого десятилетия и в трех четвертях старше 80 лет. FXTAS может встречаться и у некоторых женщин — носительниц премутации. Особенности фенотипических проявлений синдрома ломкой Х-хромосомы: Лечение синдрома ломкой Х-хромосомыК настоящему времени никакого патогенетического лечения при синдроме ломкой Х-хромосомы нет. Помощь направлена на обучение и фармакологическое лечение поведенческих проблем. Риски наследования синдрома ломкой Х-хромосомыРиск того, что женщина с премутацией будет иметь больного ребенка, определяется размером премутации, полом плода и семейным анамнезом. Эмпирически риск для носителя перестройки иметь больного ребенка может достигать 50% для каждого мальчика и 25% для каждой девочки, но зависит от размера премутации. На основе анализа сравнительно небольшого количества матерей-носительниц известно, что риск повторения может снижаться, если премутация уменьшается со 100 до 59 повторов. Пренатальная диагностика доступна за счет использования ДНК плода из ворсин хориона или амниоцитов. Пример синдрома ломкой Х-хромосомы. Р.Л., 7-летний мальчик, направлен в клинику педиатрии в связи с умственной задержкой и гиперактивностью. Он не смог посещать детский сад, поскольку был агрессивным, не в состоянии выполнять задания, имел бедные речевые и двигательные навыки. Несмотря на задержанное развитие, он не потерял основных этапов: сидел к 10-11 мес, ходить начал в 20 мес, говорил два или три ясных слова в 24 мес. В остальном ребенок здоров. Его мать и тетя по матери имели небольшие проблемы обучения в детстве, дядя по матери умственно задержан. Данные медицинского осмотра в норме, за исключением гиперактивности. Врач рекомендовал несколько тестов, включая кариотипирование, функциональные исследования щитовидной железы и ДНК-анализ на синдром ломкой Х-хромосомы. Анализ гена FMR1 методом блот-гибридизации по Саузерну соответствовал синдрому ломкой Х-хромосомы. – Также рекомендуем “Недостаточность глюкозо-6-фосфат дегидрогеназы (Г6ФД): причины, диагностика, лечение” Оглавление темы “Генетические болезни”:
|
Источник
Елена Шведкина об одном из самых распространенных генетических заболеваний — больные жалуются на бесплодие, эректильную дисфункцию, гинекомастию и остеопороз
Синдром Клайнфельтера — генетическое заболевание, характеризующееся дополнительной женской половой хромосомой Х (одной или даже несколькими) в мужском кариотипе ХY. При этом в мужских половых железах — яичках — образуется недостаточно половых гормонов.
Как известно, генетический набор человека насчитывает 46 хромосом, из которых 22 пары называются соматическими, а 23‑я пара — половая. Женщины имеют пару половых хромосом ХХ, а мужчины — ХY. Для синдрома Клайнфельтера обязательно наличие мужской Y-хромосомы, поэтому, несмотря на дополнительные Х-хромосомы, пациенты всегда являются мужчинами.
Классификация: виды кариотипов при синдроме Клайнфельтера
По количеству дополнительных Х-хромосом различают следующие варианты синдрома Клайнфельтера:
- 47,ХХY — наиболее часто встречающийся
- 48,ХХХY
- 49,ХХХХY
Кроме того, к синдрому Клайнфельтера также относят мужские кариотипы, включающие, помимо дополнительных Х-хромосом, дополнительную Y-хромосому — 48,ХХYY. И, наконец, среди пациентов с этим синдромом встречаются лица с мозаичным кариотипом 46,ХY/47,ХХY (то есть часть клеток имеет нормальный хромосомный набор).
История открытия синдрома
Синдром получил свое название в честь Гарри Клайнфельтера — врача, в 1942 году впервые описавшего клиническую картину болезни. Клайнфельтер с коллегами опубликовали отчет об обследовании 9 мужчин, объединенных общими симптомами, такими как слабое оволосение тела, евнухоидный тип телосложения, высокий рост и уменьшенные в размерах яички. Позднее, в 1956 г., генетики Планкетт и Барр (Е. R. Plankett, М. L. Barr) обнаружили у мужчин с синдромом Клайнфельтера тельца полового хроматина в ядрах клеток слизистой оболочки полости рта, а в 1959 году Полани и Форд (P. E. Polanyi, S. E. Ford) с сотрудниками показали, что у больных в хромосомном наборе имеется лишняя Х-хромосома.
Активные исследования данной патологии велись в 70‑х годах в США. Тогда всех новорожденных мальчиков подвергали кариотипированию, в результате чего удалось достоверно выявить распространенность и генетические особенности синдрома Клайнфельтера.
Любопытно, что мыши также могут иметь синдром трисомии по половым хромосомам XXY, что позволяет эффективно использовать их в качестве моделей для исследования синдрома Клайнфельтера.
Распространенность заболевания
Синдром Клайнфельтера является одним из наиболее распространенных генетических заболеваний: на каждые 500 новорождённых мальчиков приходится 1 ребёнок с данной патологией.
Кроме того, синдром Клайнфельтера — третья по распространенности эндокринная патология у мужчин (после сахарного диабета и патологии щитовидной железы) и наиболее частая причина врожденного нарушения репродуктивной функции у мужчин.
На сегодняшний день около половины случаев синдрома Клайнфельтера остаются нераспознанными. Часто такие пациенты обращаются за помощью по поводу бесплодия, эректильной дисфункции, гинекомастии, остеопороза, анемии и пр. без установленного ранее диагноза.
Этиология и причины нарушения
Синдром Клайнфельтера относится к генетическим заболеваниям, не передающимся по наследству, поскольку больные, за редким исключением, бесплодны. Патология, как правило, возникает в результате нарушения расхождения хромосом на ранних стадиях формирования яйцеклеток и сперматозоидов. При этом синдром Клайнфельтера, возникающий за счет нарушения в женских половых клетках, встречается в три раза чаще. Мозаичные формы обусловлены патологией деления клеток на ранних стадиях эмбриогенеза, поэтому часть клеток у таких пациентов имеет нормальный кариотип. Причины нерасхождения половых хромосом и нарушения деления клеток на самых ранних стадиях эмбриогенеза до сих пор малоизучены. В отличие от других хромосомных заболеваний, влияние возраста родителей отсутствует или выражено незначительно.
Ранние признаки
В отличие от большинства заболеваний, связанных с нарушением количества хромосом, внутриутробное развитие детей с синдромом Клайнфельтера проходит нормально, склонности к преждевременному прерыванию беременности не наблюдается. Так что в младенческом и раннем детском возрасте заподозрить патологию практически невозможно. Более того, клинические признаки классического синдрома Клайнфельтера проявляются, как правило, только в подростковом периоде. Однако есть симптомы, которые позволяют заподозрить наличие синдрома Клайнфельтера в препубертатном периоде:
- высокий рост (пик прибавки роста приходится на период между 5–8 годами);
- длинные ноги (непропорциональное телосложение);
- высокая талия.
У части пациентов наблюдается некоторая задержка в развитии речи.
В подростковом возрасте синдром часто проявляется гинекомастией, которая при данной патологии имеет вид двустороннего симметричного безболезненного увеличения грудных желез. Так как такого рода гинекомастия часто наблюдается у совершенно здоровых подростков, этот симптом часто остается без внимания. В норме подростковая гинекомастия бесследно исчезает в течение нескольких лет, у пациентов же с синдромом Клайнфельтера обратной инволюции грудных желез не происходит. В некоторых случаях гинекомастия может не развиваться вовсе, и тогда патология проявляется признаками андрогенной недостаточности уже в постпубертатный период.
Симптомы андрогенной недостаточности при синдроме Клайнфельтера
Андрогенная недостаточность при синдроме Клайнфельтера связана с постепенной атрофией яичек, что приводит к снижению синтеза тестостерона. Степень недостаточности андрогенов резко варьирует.
В первую очередь обращают на себя внимание внешние признаки гипогонадизма:
- скудная растительность на лице или же полное ее отсутствие;
- рост волос на лобке по женскому типу;
- волосы на груди и других частях тела отсутствуют;
- маленький объем яичек (2–4 мл) и их плотная консистенция (патогномоничный признак).
Поскольку дегенерация половых желез, как правило, развивается в постпубертатный период, у большинства пациентов размеры мужских половых органов, за исключением яичек, соответствуют возрастным нормам.
Пациенты могут жаловаться на ослабление либидо и снижение потенции. У многих мужчин с синдромом Клайнфельтера половое влечение вовсе не возникает, а некоторые — напротив, заводят семью и живут нормальной половой жизнью. Наиболее постоянный признак патологии — бесплодие, именно оно чаще всего становится причиной обращения таких пациентов к врачу. У 10 % мужчин с азооспемией обнаруживают синдром Клайнфельтера.
Всем пациентам с нарушениями сперматогенеза необходимо определять кариотип для исключения или подтверждения диагноза синдрома Клайнфельтера.
Недостаток андрогенов приводит к развитию остеопороза, анемии и слабости скелетной мускулатуры. У трети больных можно наблюдать варикозное расширение вен голеней.
Андрогены влияют на обмен веществ, поэтому больные с синдромом Клайнфельтера склонны к ожирению, нарушению толерантности к глюкозе и сахарному диабету второго типа.
Доказана предрасположенность таких пациентов к аутоиммунным заболеваниям (ревматоидный артрит, системная красная волчанка, аутоиммунные заболевания щитовидной железы и другие).
Психологические особенности
Коэффициент интеллекта у больных с классическим синдромом Клайнфельтера варьирует от значений ниже среднего до показателей, значительно превышающих средний уровень. Однако во всех случаях отмечается диспропорция между общим уровнем интеллекта и вербальными способностями, так что нередко пациенты с достаточно высоким IQ испытывают трудности при восприятии больших объемов материала на слух, а также при построении фраз, содержащих сложные грамматические конструкции. Такие особенности причиняют пациентам много неприятностей в период обучения и нередко продолжают сказываться на профессиональной деятельности.
Данные о психологических особенностях больных с синдромом Клайнфельтера достаточно противоречивы, однако большинство специалистов оценивают пациентов как скромных, робких людей с несколько заниженной самооценкой и повышенной чувствительностью. Есть данные, свидетельствующие о склонности пациентов с синдромом Клайнфельтера к гомосексуализму, алкоголизму и наркомании. Сложно сказать, вызваны ли особенности психики у таких больных непосредственным влиянием хромосомной аномалии, или же это реакция на проблемы в сексуальной сфере.
В отношении разных цитогенетических вариантов синдрома Клайнфельтера справедливо правило, что с увеличением количества дополнительных Х-хромосом увеличивается количество и выраженность патологических симптомов.
Диагностика синдрома Клайнфельтера
Во многих странах синдром Клайнфельтера часто диагностируется ещё до рождения ребёнка, так как многие женщины позднего детородного возраста, в связи с высоким риском генетических дефектов у будущего потомства, используют пренатальную генетическую диагностику плода. Нередко пренатальное выявление синдрома Клайнфельтера является поводом для прерывания беременности, в том числе и по рекомендации врачей. В России анализ кариотипа будущего ребёнка проводится крайне редко.
При подозрении на синдром Клайнфельтера проводят лабораторный анализ крови для определения уровня мужских половых гормонов. Необходима дифференциальная диагностика с другими заболеваниями, протекающими с проявлениями андрогенной недостаточности. Точный диагноз синдрома Клайнфельтера ставят на основании изучения кариотипа (набора хромосом) больного.
Исследования, необходимые для подтверждения диагноза
Анализы | Результаты |
Кариотип | 47,ХХY (80 % случаев) |
Концентрация ЛГ, ФСГ | Повышена, особенно ФСГ |
Концентрация общего тестостерона | Чаще снижена (в некоторых случаях нормальная за счет повышения секс-стероид-связывающего глобулина СССГ или на начальной стадии развития заболевания) |
У всех мужчин с резко повышенными концентрациями гонадотропинов необходимо исключить синдром Клайнфельтера, так как нередко первый лабораторный признак этой генетической патологии — повышение в крови концентрации гонадотропинов при нормальном содержании общего тестостерона.
Синдром Клайнфельтера необходимо дифференцировать от других форм первичного гипогонадизма. В любом случае при повышении уровня ФСГ в крови необходимо определение кариотипа для исключения в первую очередь синдрома Клайнфельтера.
Лечение
Цели лечения синдрома Клайнфельтера:
- Восстановление нормального содержания тестостерона
- Восстановление сексуальной функции
- Ликвидация метаболических нарушений
При клинически выраженной патологии необходима пожизненная заместительная терапия препаратами тестостерона. Адекватная терапия позволяет не только улучшить внешний вид и общее самочувствие больного, но и вернуть способность к нормальной половой жизни. Кроме того, заместительная терапия предупреждает развитие остеопороза, купирует мышечную слабость. В юном возрасте лечение необходимо начинать сразу же после постановки диагноза. При синдроме Клайнфельтера лучше использовать препараты тестостерона длительного действия:
- смесь эфиров тестостерона в виде масляного раствора, инъекции которого необходимо делать 2–3 раза в месяц;
- тестостерона ундеканоат в виде масляного раствора — препарат-депо с замедленным высвобождением действующего вещества — инъекции 1 раз в 3 месяца.
Гормонолечение при наличии Х хромосомы у мужчин должно носить постоянный характер. Дозу препарата подбирают индивидуально под контролем уровня тестостерона и ЛГ в сыворотке крови.
Уже развившаяся гинекомастия при синдроме Клайнфельтера не подвергается инволюции даже в случае адекватного лечения, поэтому часто приходится прибегать к хирургической коррекции (мастэктомии).
Для профилактики таких сопутствующих заболеваний, как ожирение и сахарный диабет второго типа, больным рекомендуют придерживаться диеты и следить за собственным весом.
Мониторинг пациентов с синдромом Клайнфельтера следует осуществлять не реже 1 раза в 6–12 месяцев. Он должен включать следующие исследования:
- общий анализ крови для оценки уровня гемоглобина и гематокрита;
- гормональный анализ крови, включающий определение тестостерона и ЛГ (проводится на фоне лекарственной терапии за 1–2 дня до очередной инъекции тестостерона);
- денситометрию (всем пациентам, у которых на момент постановки диагноза были обнаружены остеопения или остеопороз).
Внедрение интрацитоплазматической инъекции сперматозоида в яйцеклетку (ИКСИ) и данные о возможности присутствия зародышевых клеток в яичках у пациентов с синдромом Клайнфельтера предопределили применение метода искусственного оплодотворения для данной категории пациентов, некоторые попытки были удачными.
Прогноз
Прогноз для жизни и трудовой деятельности у пациентов с классическим синдромом Клайнфельтера — в целом благоприятен. Ранняя заместительная терапия, психологическая работа с пациентами и их родителями позволяют больным полностью адаптироваться в современном обществе.
Источник