
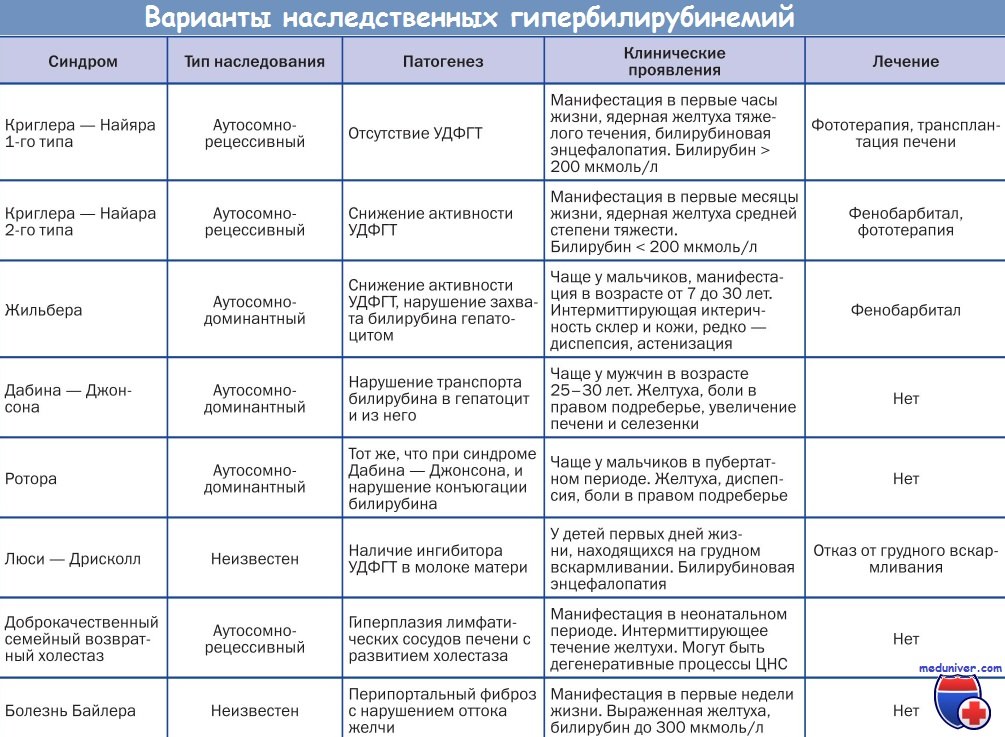
Синдромы жильбера криглера найяра дабина джонсона

Синдром Жильбера— Леребулле – семейная гипербилирубинемия. Синдром Криглера — Найяра и Люцея — Ариаса — Дрисколла
Синдром Жильбера— Леребулле, семейная гипербилирубинемия, связан с нарушением функции ферментов, обеспечивающих перенос непрямого билирубина из крови в печеночную клетку. Очевидно, происходит в какой-то степени и снижение процессов конъюнгации билирубина. Врожденное заболевание с аутосомно-доминантным типом наследования. Чаще болеют мальчики или взрослые мужчины, выявление болезни в периоде новорожденности маловероятно. Морфологические изменения в печени либо отсутствуют (у 0,25 больных), либо носят неспецифический характер.
Находят отложение липофусцина или жировую дистрофию гепатоцитов. Звездчатые эндотелиоциты образуют небольшие скопления.
При электронно-микроскопическом исследовании обнаруживается уменьшение числа и деформация микроворсинок на синусоидальном полюсе гепатоцитов.
Синдром Криглера — Найяра — тяжелое заболевание, связанное с врожденным отсутствием ферментов, участвующих в конъюгации билирубина. Наследуется аутосомно-рецессивно, встречается редко. Выявляется заболевание в периоде иоворожденности тяжелой желтухой, связанной с высоким (684 мкмоль/л — 40 мг% и более) содержанием непрямого билирубина.
Небольшое количество его выделяется железами кишечника, в связи с чем кал окрашен в бледно-желтый цвет. Обычно развивается смертельная билирубиновая энцефалопатия. Описаны единичные случаи, когда больные, несмотря на желтуху доживают до зрелого возраста.
Некоторые данные и, в частности, наличие обоих заболеваний в одних и тех же семьях свидетельствуют о близости синдромов Жильбера — Леребулле и Криглера — Найяра. Последний, по-видимому, является крайним вариантом первого.
Печень несколько увеличена лишь в редких случаях. Микроскопически выявляются желчные «тромбы» в канальцах, у части больных небольшой перипортальный фиброз или значительная жировая дистрофия гепатоцитов. При электронно-микроскопическом исследовании обнаруживается повреждение мембраны гепатоцита на синусоидальном полюсе с уменьшением числа микроворсинок и выходом органелл в перикатшллярные пространства (Диссе). Имеется также гиперплазия цитоплазматической сети, отложение желчного пигмента в гепатоцитах и эндотелиоцитах. Липофусцин в гепатоцитах не содержится.
Изменения мозга заключаются в ядерной желтухе с окрашивании гиппокамповой извилины, ядер ствола мозга и мозжечка. Имеются также очаги некроза в миокарде и скелетных мышцах, эрозии слизистой оболочки желудка и кишечника.
Преходящая семейная желтуха Люцея — Ариаса—Дрисколла, обменные или лекарственные гипербилирубинемии, проявляются в периоде иоворожденности (иногда на 7—9-е сутки) интенсивной желтухой с наличием в крови непрямого билирубина.
Нарушение конъюгации билирубина происходит при синдроме Люцея — Ариаса — Дрисколла в результате того, что на ферментные системы связывания билирубина Действует ингибитор, проникающий через плаценту от беременной женщины и являющийся, очевидно, прогестероном.
Аналогичное действие вызывают стероиды, в частности 3-а-20-бета-прегнандиол вырабатываемые в организме некоторых родильниц и поступающие в организм новорожденного с молоком. Прекращение кормления ребенка молоком матери ведет в этих случаях к быстрой нормализации билирубина, но возможно и тяжелое течение с развитием билирубиновой энцефалопатии. Как и при других желтухах, связанных с нарушением конъюгации билирубина, при синдроме Люцея — Ариаса — Дрисколла отсутствуют признаки гемолиза и грубые поражения печели. Отсутствует и плейохромия (интенсивная окраска) желчи и кала.
Ранняя желтуха новорожденного может быть также обусловлена поступлением через плаценту во время родов из организма беременной женщины эстрогенов, метаболитов, тормонов надпочечников и др. Связывание этих веществ происходит так же, как и непрямого билирубина. Перегрузка ферментных систем приводит к задержке выделения билирубина.
После рождения аналогичное конкурентное действие могут оказать многие лекарственные вещества (барбитураты, салицилаты, сульфаниламиды и др.).
Для конъюгации билирубина необходима глюкоза (в виде уридиндифосфатглюкозы), поэтому состояния, сопровождающиеся дефицитом глюкозы в организме новорожденного и гипогликемией, могут осложниться задержкой непрямого билирубина.
– Также рекомендуем “Физиологическая желтуха новорожденных. Синдромы Дабина — Джонсона и Ротора”
Оглавление темы “Врожденные формы желтухи у новорожденных”:
1. Послеродовая желтушная форма гемолитической болезни. Анемическая форма гемолитической болезни
2. Плацента при гемолитической болезни. Билирубиновая энцефалопатия
3. Ядерная желтуха. Признаки ядерной желтухи у плода
4. Послежелтушная энцефалопатия. Наследственный микросфероцитоз – болезнь Минковского-Шоффара
5. Наследственный стоматоцитоз. Дефицит глюкозо-6-фосфатдегидрогеназы плода
6. Дефицит пируваткиназы у плода. Гемоглобинозы
7. Гемоглобиноз S. Талассемия у новорожденных
8. Классификация желтух новорожденных. Синдром Израельса
9. Синдром Жильбера— Леребулле – семейная гипербилирубинемия. Синдром Криглера — Найяра и Люцея — Ариаса — Дрисколла
10. Физиологическая желтуха новорожденных. Синдромы Дабина — Джонсона и Ротора
Источник

Синдром Дабина-Джонсона — это хроническое наследственное заболевание, которое характеризуется нарушением выделения билирубина из гепатоцитов в желчь. Основное клиническое проявление — интермиттирующая желтуха. Для болезни также характерны диспепсические расстройства, снижение аппетита и ухудшение общего самочувствия. Диагностика синдрома Дабина-Джонсона включает биохимические анализы крови и мочи, бромсульфалеиновую пробу, инструментальные методики (УЗИ, лапароскопию, биопсию печени). Лечение предполагает коррекцию образа жизни, назначение щадящей диеты. При необходимости используют желчегонные препараты и другие лекарственные средства.
Общие сведения
Синдром имеет несколько синонимов — энзимопатическая желтуха, генетически обусловленный пигментный гепатоз. Нозологическая форма названа в честь двух американских ученых — И.Н. Дабина и Ф.Б. Джонсона, описавших синдром в 1954 году. Это редкое заболевание, в 70% проявляющееся в молодом возрасте. В основном синдром Дабина-Джонсона наблюдается у жителей Среднего Востока. Наибольшая частота встречаемости среди иранских евреев — 1 случай на 1300 населения. У 60% больных патология сопровождается снижением активности факторов коагуляции и кровотечениями. Заболеваемость не зависит от пола.

Причины
Синдром имеет генетическую природу, характеризуется аутосомно-рецессивным путем наследования. Среди родственников болезнь может проявляться как у мужчин, так и у женщин, повторяется через 1-2 поколения. Наследственный дефект представлен мутацией в нуклеотидной последовательности, обеспечивающей кодирование белка MRP2. Этот протеин отвечает за экскрецию (выделение) конъюгированного билирубина и органических анионов в желчные ходы.
Патогенез
Вследствие дисфункции АТФ-зависимой транспортной системы канальцев желчь не проходит в желчные капилляры, из-за чего билирубин накапливается в печеночных тканях. Длительно существующий синдром сопровождается обратными биохимическими реакциями: избыточное количество прямого билирубина освобождается от глюкуроновой кислоты. Полученный непрямой билирубин также проникает в кровоток, обуславливает токсическое действие на нервную систему.
Нарушается выделение активных метаболитов эпинефрина (триптофана, тирозина). Как следствие, в печени накапливаются меланиноподобные пигменты, расположенные в лизосомах гепатоцитов. При макроскопическом исследовании в печеночной паренхиме видны множественные темные пятна — так называемая «шоколадная печень». При исследовании образцов под микроскопом наблюдаются скопления пигментных зерен большей частью в центре долек.
Симптомы
Обычно манифестация синдрома Дабина-Джонсона происходит в 20-30-летнем возрасте, хотя первые характерные признаки появляются уже у подростков. Крайне редко заболевание выявляется у детей. У женщин с бессимптомным течением энзимопатической желтухи клинический дебют болезни могут спровоцировать наступление беременности или прием контрацептивных средств.
Основный симптом заболевания — желтуха, не сопровождаемая кожным зудом. Сначала возникает желтушность склер и слизистых оболочек, затем кожа также приобретает желтушный оттенок. Желтуху могут усиливать изнурительные физнагрузки, стрессовые ситуации, интеркуррентные инфекции. Как правило, желтушные периоды сменяются безжелтушными, хотя у ряда пациентов иктеричность кожи сохраняется постоянно.
Синдром периодически обостряется. Пациент жалуется на сильные боли справа в подреберье, реже — в околопупочной области. Иногда боль отдает в правое плечо или лопатку. Болевой синдром может быть настолько интенсивным, что напоминает печеночную колику. Одновременно с болью беспокоят тошнота, горечь во рту. Изредка бывает рвота, которая не приносит облегчения. Общая интоксикация проявляется повышенной утомляемостью, сонливостью, снижением аппетита.
Осложнения
Синдром Дабина-Джонсона отличается доброкачественным течением и при правильном лечении не вызывает неприятных последствий. Частое осложнение при длительно протекающем заболевании — воспалительные процессы в желчном пузыре и протоках. Воспаление может переходить и на печеночную паренхиму, вызывая холестатический гепатит. При наличии этих патологий желтуха у больных синдромом Дабина-Джонсона становится постоянной.
При присоединении бактериальной инфекции может возникать эмпиема желчного пузыря и гнойный холангит, которые при неблагоприятных условиях трансформируются в ограниченный перитонит. У пожилых людей наследственный пигментный синдром провоцирует появление фиброзных изменений в печеночных дольках. В результате снижается функциональная активность печени. Вследствие нарушения синтеза факторов свертывания крови возрастает риск кровотечений.
Диагностика
При физикальном обследовании пациентов врач-гастроэнтеролог или гепатолог пальпирует выступающий на несколько сантиметров из-под реберной дуги край печени. Синдром дифференцируют с болезнью Ротора, гепатитами, раковыми метастазами в печень. Симптомы поражения желчного пузыря (Мюсси, Ортнера, Кера) отрицательные. Для подтверждения заболевания используют следующие лабораторные и инструментальные исследования:
- Анализы крови. Основным диагностическим критерием синдрома является повышение уровня общего билирубина более 85 мкмоль/л, при этом массовая доля прямого билирубина превышает 15%. При выполнении коагулограммы у 60% пациентов выявляют снижение активности протромбина и уменьшение протромбинового времени.
- Анализ мочи. В моче обнаруживают повышенный уровень билирубина. Для дифференциальной диагностики с близким по клинической симптоматике синдромом Ротора оценивают соотношение копропорфиринов I и III типа в моче. При гепатозе Дабина-Джонсона количество копропорфирина 1 типа составляет 80%, а 3 типа — 20%.
- Бромсульфалеиновая проба. Анализ наиболее чувствителен для оценки печеночных функций. При синдроме Дабина-Джонсона спустя 45 минут концентрация в крови специального красителя, который предварительно ввели внутривенно, составляет более 6%. Такая проба считается положительной и указывает на снижение поглотительно-выделительной функции печени.
- УЗИ органов брюшной полости. При сонографии синдром проявляется увеличением печени на 1-2 см, у пациентов среднего и пожилого возраста нередко обнаруживают очаги фиброза. Размеры и контуры желчного пузыря не изменены, конкременты не визуализируются. Характерно одновременное увеличение селезенки.
- Инвазивные методы. В сомнительных ситуациях показана диагностическая лапароскопия, которая позволяет врачу осмотреть наружную поверхность печени и выявить характерные коричневые пятна. Чтобы подтвердить диагноз, назначается чрескожная биопсия печеночной ткани с последующим осмотром биоптатов под электронным микроскопом.
Лечение синдрома Дабина-Джонсона
Этиопатогенетическое лечение заболевания не разработано. Главная роль в предупреждении ухудшений состояния больных принадлежит немедикаментозным мероприятиям. Чтобы предотвратить обострения синдрома Дабина-Джонсона, пациентам рекомендуют по возможности избегать физического переутомления и стрессов. Основные направления лечения, применяемые в современной гастроэнтерологии для улучшения качества жизни человека:
- Диета. В рационе питания ограничивают потребление тугоплавких жиров и продуктов, содержащих консерванты. Полностью исключают употребление спиртных напитков. Показана витаминизированная диета с достаточной калорийностью.
- Препараты с желчегонным эффектом. Назначаются синтетические или растительные лекарственные средства, которые либо увеличивают концентрацию желчных кислот, либо повышают содержание водного компонента желчи. Специалисты отдают предпочтение мягким растительным препаратам.
- Санация очагов инфекции. Проводится выявление и лечение самых распространенных хронических источников инфекции — кариеса зубов и хронического тонзиллита. При обнаружении сопутствующей патологии желчевыводящих путей подбирается соответствующая патогенетическая терапия.
Прогноз и профилактика
Наличие болезни Дабина-Джонсона не влияет на продолжительность жизни и работоспособность, поэтому прогноз благоприятный. Синдром не прогрессирует. При соблюдении врачебных рекомендаций пациенты чувствуют себя хорошо. Больным необходимо воздерживаться от алкоголя и приема контрацептивов. Специфическая профилактика не разработана. Семьям с синдромом Дабина-Джонсона перед планированием беременности необходимо проконсультироваться с врачом-генетиком.
Литература 1. Этапы диагностического поиска при синдроме желтухи: лекции/ Л.Н. Бобро, Л.М. Пасиешвили. — 2010. 2. Клинические особенности течение синдрома Дабина-Джонсона у детей/ В.С. Березенко, М.Б. Диба, Ю.П. Резников// Современная педиатрия. — 2018. 3. Доброкачественные гипербилирубинемии: всегда ли они доброкачественные?/ Н.Б. Губериц// Новости медицины и фармации. — 2011. 4. Пигментные гепатозы: клинические особенности, пункционная биопсия, электронная микроскопия, диагноз, прогноз/ С.Д. Подымова// Практическая гастроэнтерология. — 2018. | Код МКБ-10 E80.6 |
Синдром Дабина-Джонсона – лечение в Москве
Источник

Синдром Криглера-Найяра – генетическое заболевание из класса ферментопатий, характеризующееся нарушением одного из звеньев процесса обезвреживания и выведения билирубина – конъюгации. Симптомами этого состояния являются желтуха печеночного генеза и тяжелые неврологические нарушения, которые могут привести к летальному исходу еще в младенческом возрасте. Диагностика синдрома Криглера-Найяра производится посредством биохимических проб и определения уровня неконъюгированного билирубина в плазме крови, а также молекулярно-генетическими методиками. Специфического лечения заболевания не существует (за исключением трансплантации печени), терапия сводится к увеличению разрушения и элиминации билирубина из организма (гемосорбция, фототерапия, плазмаферез, прием барбитуратов).
Общие сведения
Синдром Криглера-Найяра – тяжелое генетическое заболевание, характеризующееся нарушением связывания билирубина с глюкуроновой кислотой, что является ключевым этапом его обезвреживания и выведения из организма. Впервые это заболевание было описано в 1952 году двумя американскими педиатрами – Джоном Криглером и Виктором Найяром. Дальнейшее изучение синдрома Криглера-Найяра показало, что это состояние имеет генетическую природу и аутосомно-рецессивный характер наследования, кроме того, удалось выявить две клинические разновидности данной патологии. Заболевание достаточно редкое, поэтому точные цифры встречаемости не определены – большинство исследователей полагает, что она находится на уровне 1:1 000 000. Половое распределение больных синдромом Криглера-Найяра не имеет особенностей, заболевание с одинаковой частотой поражает как мальчиков, так и девочек. В лечении этого состояния крайне важна ранняя (в идеале – пренатальная) диагностика, так как от своевременности начатой терапии очень сильно зависят прогноз заболевания и качество жизни больного.
Синдром Криглера-Найяра
Причины синдрома Криглера-Найяра
Синдром Криглера-Найяра относят к классу ферментопатий (по другой классификации – к группе неконъюгированных гипербилирубинемий), причина этого заболевания кроется в недостаточности уридиндифосфатглюкуронидазы 1, функцией которой является связывание билирубина с двумя молекулами глюкуроновой кислоты. В итоге этого биохимического процесса билирубин становится способным растворяться в воде, выводиться в составе желчи и, главное, значительно падает его токсичность. При синдроме Криглера-Найяра этот процесс резко замедлен или не происходит совсем, вследствие чего возникает задержка элиминации билирубина из организма и его накопление. Билирубин обладает выраженной нейротоксичностью, при повышении концентрации в крови это вещество начинает откладываться в тканях кожных покровов и слизистых оболочек, приводя к развитию желтухи. Когда концентрация билирубина превышает определенный порог, соединение начинает проникать через гематоэнцефалический барьер в головной мозг, приводя к тяжелой энцефалопатии (особенно повреждаются базальные ядра). При отсутствии лечения больные синдромом Криглера-Найяра погибают от многочисленных неврологических расстройств и нарастающей печеночной комы.
Причиной низкой активности уридиндифосфатглюкуронидазы являются мутации гена UGT1A1, который располагается на 2-й хромосоме, отвечает за аминокислотную последовательность и выделение этого фермента. Помимо синдрома Криглера-Найяра дефекты этого гена могут приводить к ряду других нарушений билирубинового обмена наследственного характера – синдрому Жильбера, транзиторной неонатальной билирубинемии семейного типа. Механизм наследования мутаций гена UGT1A1 при синдроме Криглера-Найяра аутосомно-рецессивный. При этом описано несколько вариантов возможного повреждения этого гена, которые приводят к разному течению данного заболевания.
Классификация и симптомы синдрома Криглера-Найяра
В настоящее время описаны две основные клинические формы синдрома Криглера-Найяра, в основном различающиеся между собой тяжестью проявлений и прогнозом заболевания. Это обусловлено типом генетического дефекта в UGT1A1. Первый тип заболевания (СКН-1) вызывается миссенс-мутациями, приводящими к появлению неполноценного фермента, имеющего сигнальную последовательность аминокислот, характерную для подвергающихся внутриклеточной утилизации белков. Таким образом, при этой форме дефект гена поражает кодирующие участки (экзоны), что вызывает развитие патологии у гомозигот. Вскоре после своего образования уридиндифосфатглюкуронидаза 1 разрушается и конъюгации билирубина не происходит совсем.
Синдром Криглера-Найяра 1-го типа характеризуется тяжелым и стремительным течением – первые признаки гипербилирубинемии в виде желтухи обнаруживаются уже через несколько часов после рождения. Со временем к ним присоединяются неврологические нарушения – нистагм, судорожные приступы, иногда возникает опистотонус. Желтуха сохраняется на протяжении всей жизни ребенка, его умственное развитие резко отстает от такового у сверстников, симптомы заболевания неуклонно нарастают даже при интенсивном лечении. Обычно больные синдромом Криглера-Найяра 1-го типа умирают на протяжении первого года жизни из-за интоксикации билирубином и поражения базальных подкорковых ядер (ядерная энцефалопатия).
Причиной синдрома Криглера-Найяра 2-го типа также являются миссенс-мутации гена UGT1A1, однако они могут возникать как в кодирующей последовательности (экзонах), так и в промоторе – участке, отвечающем за экспрессию данного гена. У большинства больных СКН-2 наблюдается наличие на одной хромосоме дефекта экзона, на другой – промотора, то есть, такие лица являются компаунд-гетерозиготами. Результатом нарушения является продукция дефектной формы фермента уридиндифосфатглюкуронидазы, которая не разрушается, но имеет пониженную (на уровне 20-25% от нормы) функциональную активность. Поэтому синдром Криглера-Найяра 2-го типа характеризуется менее тяжелой клинической картиной и более благоприятным прогнозом.
В первые месяцы и даже годы жизни больных синдром Криглера-Найяра этого типа нередко проявляется только незначительной желтухой, при отсутствии лечения к подростковому периоду могут развиваться неврологические отклонения. В ряде случаев, особенно при правильно назначенных терапевтических мероприятиях, никаких нарушений со стороны центральной нервной системы не возникает вовсе. Проявления желтухи различной степени выраженности у больных синдромом Криглера-Найяра 2-го типа могут сохраняться на протяжении всей жизни и нередко расцениваются как индикатор осложнений и ухудшения состояния пациента. С возрастом иногда появляется нистагм, могут регистрироваться судорожные припадки, однако течение и выраженность симптомов заболевания всецело зависят от качества лечения и выполнения рекомендаций специалистов.
Диагностика синдрома Криглера-Найяра
Диагностика синдрома Криглера-Найяра производится на основании данных общего осмотра ребенка, биохимических исследований крови, желчи и мочи, молекулярно-генетических анализов. При осмотре выявляется желтуха, возникшая в первые часы (при СКН-1) или месяцы (СКН-2) жизни, признаки неврологических нарушений (опистотонус, нистагм, длительное сохранение транзиторных рефлексов). У больных 2-м типом синдрома Криглера-Найяра неврологические расстройства могут регистрироваться во взрослом возрасте, тогда как у детей наблюдается только желтуха. Также с возрастом могут присоединяться такие проявления, как нейросенсорная глухота или хореоатетоз.
При биохимическом исследовании крови выявляется выраженная непрямая гипербилирубинемия (вплоть 200-350 мкмоль/л), отсутствие (при синдроме Криглера-Найяра 1-го типа) или резкое снижение концентрации прямого билирубина. Конъюгированная фракция этого соединения отсутствует в желчи при СКН-1 и присутствует в незначительных количествах при СКН-2. Фенобарбиталовая проба при синдроме Криглера-Найяра положительна только в случае наличия уридиндифосфатглюкуронидазы, то есть при СКН-2. Изучение концентрации неконъюгированного билирубина в моче показывает его увеличение. Молекулярно-генетическая диагностика синдрома Криглера-Найяра производится врачом-генетиком – он совершает прямое секвенирование последовательности гена UGT1A1 с целью выявления мутаций. При отягощенной по этому заболеванию наследственности у родителей может осуществляться пренатальная диагностика патологии. Дифференциальный диагноз следует проводить с обычной транзиторной желтухой новорожденных и синдромом Жильбера.
Лечение синдрома Криглера-Найяра
Специфического или этиотропного лечения синдрома Криглера-Найяра на сегодняшний день не существует, все терапевтические мероприятия назначаются для ускорения распада билирубина, его выведения из организма и защиты ЦНС. Особых отличий в терапии 1-го или 2-го типа заболевания нет (за исключением активизации микросомального окисления барбитуратами, которая не производится при 1-м типе), однако при СКН-1 лечение лишь незначительно оттягивает наступление летального исхода. Самым радикальным методом лечения синдрома Криглера-Найяра в настоящее время является операция по аллотрансплантации печени от родственника или генетически сходного донора – в этом органе происходит образование уридиндифосфатглюкуронидазы.
Синдром Криглера-Найяра 2-го типа лечат назначением умеренных доз барбитуратов для активации окисления билирубина и увеличения образования нужного фермента. Кроме того, показаны плазмаферез, гемосорбция, заместительное переливание крови – все эти процедуры направлены на удаление неконъюгированного билирубина из организма. Неплохие результаты у больных синдромом Криглера-Найяра дает фототерапия – облучение кожных покровов приводит к частичному разрушению билирубина и освобождению рецепторов тканей для новых порций этого токсина, что снижает его концентрацию в крови. Правильный питьевой режим и повышенное потребление жидкости ускоряет выведение токсина из организма, поэтому следует избегать обезвоживания. Необходим постоянный мониторинг уровня этого вещества в плазме крови, особенно опасным считается его количество свыше 300-340 мкмоль/л – при такой концентрации билирубин становится способным проникать через гематоэнцефалический барьер.
Прогноз и профилактика
Прогноз синдрома Криглера-Найяра 1-го типа исключительно плохой – из-за полного отсутствия активности фермента уридиндифосфатглюкуронидазы 1 больные умирают на протяжении первого года жизни из-за осложнений ядерной энцефалопатии. Течение СКН-2 зависит от таких факторов, как выраженность проявлений, своевременность диагностики и начала лечения, соблюдения рекомендаций специалистов, наличия или отсутствия сопутствующих заболеваний. В большинстве случаев прогноз относительно благоприятный – больные синдромом Криглера-Найяра 2-го типа могут прожить до преклонного возраста, из характерных проявлений патологии их может беспокоить только желтуха. Профилактика этого состояния возможна только в рамках консультации генетика для родителей, имеющих отягощенную наследственность по этому заболеванию, а также при помощи пренатальной диагностики.
Источник