
Сколько телец барра при синдроме шерешевского

 Синдром женского гипогонадизма — хроническое хромосомное заболевание характеризующееся многочисленными половыми и соматическими нарушениями.
Синдром женского гипогонадизма — хроническое хромосомное заболевание характеризующееся многочисленными половыми и соматическими нарушениями.
Обусловлено отсутствием женщин второй Х-хромосомы (всего в наборе 45 хромосом).
Моносомия по Х-хромосоме возникает чаще всего в результате оплодотворения аберрантной яйцеклетки, лишенной Х-хромосомы, спермием с Х-хромосомой.
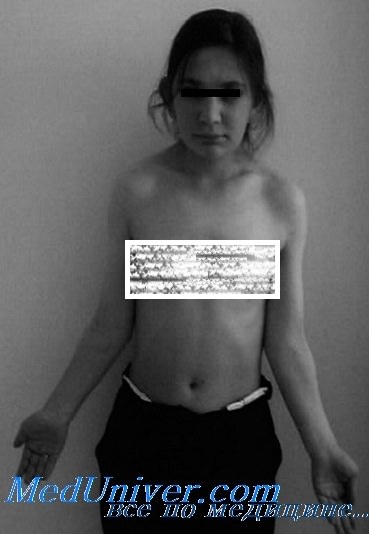
Один из важных признаков синдрома Шерешевского Тернера — низкий рост (новорожденные девочки имеют небольшую массу тела и размеры, рост взрослых —130— 140 см). Короткая шея и кожные складки от затылка к надплечьям придают больным «вид сфинкса» (рис. 59). Впубертатном возрасте отчетливо выявляется резкий половой инфантилизм — гениталии, яичники и грудные железы недоразвиты. Менструации отсутствуют. Выделение эстро по сравнению с нормой снижено в 10—12 раз. Больные не способны к деторождению. Около 10% женского бесплодия имеют в основе синдром Шерешевского Тернера. Характерны также органические изменения в виде птоза, катаракты, миопии, остеопороза, врожденных вывихов. Интеллект большинства больных близок к норме, в части случаев имеется небольшая умственная отсталость.
Рис. Половой хроматин (1) и барабанные палочки (2) в лейкоцитах крови при различном наборе хромосом.
Как и при других хронических заболеваниях, имеются изменения дерматоглифики — кожного рисунка ладоней и стоп (сгибательные складки ладоней, расположение три-радиуса и др.).
Количественные и структурные аномалии известны и по другим хромосомам (трисомия 13-й хромосомы — синдром Патау, трисомия 18-й хромосомы — синдром Эдварса и др.) и тоже сопровождаются широким спектром тяжелых нервно-психических, эндокринных и соматических нарушений умственной отсталостью, уродствами в строении скелета, атрофии мышц, поражениями нервной системы. При нарушениях набора или структурных аномалиях по первым группам хромосом (1—12) организм обычно нежизнеспособен, по остальным (за исключением трисомии 21) мало жизнеспособен, и в большинстве случаев смерть наступает внутриутробно или в первые годы жизни ребенка.
Синдром нерасхождения хромосом может возникнуть не только во время гаметогенеза, но и в начальных стадиях дробления зиготы. В последнем случае часть клеток организма будет иметь нормальный, а часть — измененный хромосомный комплекс (явление мозаицизма). Клиническая картина нарушений в этих случаях бывает менее тяжелой.
Полисомия по X-Y-хромосомам (XXX, ХХХХ, ХХХХХ, XXXY, XXXXY, XXXXYYY) тоже ведет к снижению интеллекта, инфантильности агрессивности (XYY), отклонениям в физическом развитии, причем с увеличением добавочных хромосом X и в кариотипе отмечается и увеличение изменений. Так, трисомия-Х (XXX) и дисо-мия-Y (XYY) не сопровождаются бесплодием, выраженными нарушениями умственного и физического развития. Для тетра- и пентасомии-Х (ХХХХ и ХХХХХ) и трисомии-Y (XYYY)характерны грубые изменения интеллекта, соматические аномалии, недоразвитые гениталии и т. д.
Нарушения в хромосомном наборе яйцеклетки отмечаются преимущественно у женщин в возрасте 35-40 лет, вероятно, вследствие возрастных изменений метаболизма, «перезревания» клеток. Другие причины, могущие изменять нормальный процесс мейоза: интоксикации (алкоголь, курение и др.), инфекции, особенно вирусные, облучение, нервно-психические и физические перенапряжения и т. д.
Диагностика синдрома Шерешевского Тернера, хромосомных болезней основана на клинических данных и исследовании (специальными методами) хромосомного набора — кариотипа и полового хроматина. Для определения кариотипа используют как прямые, так и непрямые методы исследования. В первом случае материал, взятый из костного мозга, лимфатических узлов или других тканей, изучают сразу же после получения. Однако прямой метод информативен только тогда, когда в материале имеется достаточное количество метафаз митоза, так как только в этой фазе хромосомы приобретают присущие им особенности строения и возможна их точная идентификация.
В настоящее время широко применяют непрямые методы исследования, когда взятую культуру (лимфоциты периферической крови и др.) помещают в питательную среду для культивирования. Продолжительность исследования зависит от скорости накопления делящихся клеток и может занять от 3 сут до 2 нед. Однако в соматических клетках человека имеются две морфологические структуры, определяющие пол — Х-хроматин (тельца Барра и барабанные палочки) у женщин и Y-хроматин у мужчин.
Для исследования полового хроматина X и полового хроматина Y берут обычно лейкоциты крови или соскоб слизистой рта. В норме в клетках женского организма при определенных способах окраски вблизи ядерной мембраны образуется интенсивно окрашиваемое тельце — половой хроматин, или тельце Барра (по имени ученого Барра, который в 1949 г. обнаружил в ядрах клеток кошек глыбку гетерохроматина, отсутствующую в клетках котов). Для выявления глыбок полового хроматина наиболее распространен экспресс-метод окраски по Сандерсу с использованием 2% раствора уксуснокислого ацетоорсеина и последующей иммерсионной микроскопией. Кроме того, выявляется еще и так называемая барабанная палочка, причем число телец хроматина (телец Барра) и барабанных палочек на единицу меньше числа Х-хромосом.
При синдроме Клайнфелтера (XXY) в клетках тела определяют тельце полового хроматина и барабанную палочку; при синдроме трисомии по хромосоме X (XXX) — два тельца полового хроматина и две барабанных палочки; тетрасомии (ХХХХ) — три тельца Барра и три барабанных палочки и т. д. (рис.). При синдроме Шерешевского — Тернера половой хроматин и барабанные палочки, наоборот, отсутствуют (вследствие наличия только одной Х-хромосомы).
Можно исследовать и Y-хроматин — интенсивно светящееся тельце (точка), обнаруживаемое в ядрах клеток у мужчин при определенном методе окраски. Определение Y-хроматина может быть осуществлено при использовании флюоро-хромных красителей (акрихина или акрихин-иприта) с последующей люминесцентной микроскопией. Число Y-телец соответствует количеству Y-хромосом в кариотипе — при синдроме XYY —два светящихся тельца, синдроме XYYY — три и т. д.
Метод определения полового хроматина быстрее и проще, чем исследование набора хромосом (кариотипа), поэтому он применяется в качестве одного из скрининг-тестов при массовых обследованиях населения для выявления людей с аномалиями половых хромосом, диагностике пола при так называемых интерсексуальных состояниях (особенно при ложном женском гермафродитизме), для пренатального установления пола и т. д. Определение Х-хроматина в комплексе с определением Y-хроматина дает возможность выявить набор половых хромосом без кариотипирования. Пренатальная диагностика наследственных заболеваний у беременных осуществляется с использованием амниоцентеза, который производится обычно на 13—18-й неделях беременности и позволяет исследовать клетки амниотической жидкости на цитогенетические и метаболические дефекты. Метод амниоцентеза дает возможность диагностировать все хромосомные болезни, а в сочетании с рентгенографией и ультрасонографией и многие пороки развития нервной системы. Показания к амниоцентезу: наличие аномалии у живорожденных или мертворожденных детей беременной женщины, беременность в возрасте старше 40 лет, инфекционные вирусные заболевания, облучение, травма или интоксикации в I триместре беременности
Статья на тему Синдром Шерешевского Тернера
Источник
Исследование полового хроматина. Синдром Шерешевского—ТернераПоловой хроматин, впервые выявленный в ядрах нервных клеток кошек (Барр, Бертрам), представляет собой X хромосому в неактивном состоянии, располагающуюся под ядерной оболочкой. Половой хроматин выявляется (положителен) только при наличии не менее двух X хромосом в кариотипе, так как одна из них всегда активна. Увеличение количества хромосом в кариотипе сопровождается увеличением числа хроматиновых телец, которое всегда меньше на одну X хромосому, при сопоставлении его с количеством X хромосом. Половой хроматин можно исследовать в ядрах эпителия слизистой оболочки полости рта, влагалища, клетках тканей, опухолях, клетках амниотической жидкости (с целью определения генетического пола плода), лейкоцитах. Наиболее удобным является метод Сандерсона — определение полового хроматина в ядрах эпителия слизистой оболочки полости рта. При помощи шпателя делают соскоб с внутренней поверхности щек и наносят его на предметное стекло. Препарат фиксируют уксуснокислым арсеином, который окрашивает ядерные структуры, и микроскопируют. Хроматиновые тельца прилегают к ядерной оболочке. При различных аномалиях полового развития исследование полового хроматина позволяет выявлять несоответствие между фенотипом и генетическим полом больных, что способствует правильной диагностике заболевания. Синдром Шерешевского—ТернераСиндром впервые описан Н. Л. Шерешевским в 1925 г. и Тернером в 1938 г., которые связывали развитие заболевания с недостаточностью функции гипофиза и половых желез. Причиной указанного выше клинического синдрома могут служить различные варианты мозаицизма 45ХО/46ХХ; 45XO/46XY; 45ХО/47ХХХ и др. Во всех случаях аномалий хромосомного комплекса имеется недостаток генетической информации, полной или частичной, обусловленный потерей второй половой хромосомы, что в свою очередь вызывает гормональный дисбаланс в организме и развитие гипогенитализма, задержку роста и других признаков, характерных для синдрома Шерешевского—Тернера. Клиническая картина синдрома Шерешевского-Тернера. Распространение заболевания среди новорожденных составляет 1:2500, в популяции 4:9166, а среди низкорослых 3:41. Другим чрезвычайно характерным клиническим признаком является половой инфантилизм, который характеризуется отсутствием вторичных половых признаков (отсутствие или слабое развитие молочных желез, скудное оволосение на лобке и под мышками), отсутствием самостоятельных менструаций. Яичники представлены соединительнотканными тяжами с участками зародышевой ткани яичника без примордиальных фолликулов. Матка гипопластична, а в некоторых случаях вместо нее обнаруживают соединительнотканные тяжи, наружные половые органы инфантильны. В отдельных случаях наблюдается гипертрофия клитора и избыточное оволосение по мужскому типу. При этом, кроме рудиментарного яичника, в брюшной полости выявляют зачатки ткани яичка, что объясняется присутствием в хромосомном наборе мужских половых хромосом наряду с женскими 45XO/46XY.
Синдром Шерешевского — Тернера характеризуется также большей или меньшей степенью выраженности врожденных пороков развития скелета и внутренних органов. Общий вид больных весьма своеобразен. Обращает на себя внимание лицо больных с обычно укороченной нижней челюстью. Часто имеют место врожденные дефекты органа зрения (косоглазие, птоз, эпикантус, различный уровень расположения глаз, цветовая слепота и т. д.). Характерными признаками являются низкое расположение ушных раковин и низкая граница роста волос на шее. На лице в туловище часто имеется большое количество родимых пятен. Очень характерным, но непостоянным признаком является наличие кожных складок на шее, идущих от головы к плечам и придающих вид «головы сфинкса». Телосложение больных обычно пропорционально, грудная клетка широкая, уплощенная в передне-заднем размере, иногда с вдавлением в области грудины. Соски молочных желез широко расставлены. При общем осмотре часто выявляются пороки развития скелета: укорочение четвертых метакарпальных и метатарзальных костей, синдактилия, деформация кисти типа Маделунга, вальгуспая девиация локтевых и коленных суставов. Окостенение эпифизарных хрящей несколько отстает от паспортного возраста, однако разница между костным и паспортным возрастом не превышает 2—3 лет. Пороки развития внутренних органов обычно выражаются в стенозе перешейка аорты, незаращении межжелудочковой перегородки и боталлова протока, стенозе легочной артерии, окклюзии почечных артерий, проявлением чего нередко является гипертония. В отдельных случаях наблюдаются пороки развития почек в виде двойных лоханок, двойных мочеточников, гипоплазии или подковообразной почки. Функциональное состояние передней доли гипофиза характеризуется увеличением секреции гонадотропных гормонов; уровень гормона роста в сыворотке крови обычно нормален или несколько повышен, что при задержке роста свидетельствует о понижении чувствительности периферических тканей к его действию. Функция щитовидной железы, по данным основного обмена, нормальна или несколько понижена. Уровень белковосвязанного йода находится в пределах нормы, а поглощение I131 щитовидной железой несколько ускорено. Клинических признаков нарушения функции щитовидной железы у больных не наблюдается. Имеются литературные данные о большой частоте аутоиммунного зоба при синдроме Шерешевского — Тернера, однако эти данные подтверждают не все исследователи. Выделение с мочой эстрогенов значительно уменьшено, а гонадотропинов— увеличено. Нарушения функции центральной нервной системы выражаются в некотором снижении интеллекта, которое выявляется у небольшой группы больных. Изменения электроэнцефалограммы выражаются в общемозговых нарушениях с акцентом патологии в мезодиэнцефальных структурах головного мозга, в основе которых лежит, по-видимому, хромосомная патология. – Также рекомендуем “Диагностика и лечение синдрома Шерешевского—Тернера. Чистый гонадальный дисгенез” Оглавление темы “Аномалии половых хромосом и желез”:
|
Источник
Синдром Х0-синдром Шерешевского—Тернера. Синдром XXY – синдром Клайнфельтера в неврологии
Синдром Х0 обусловлен нехваткой генетического материала, локализованного в Х-хромосоме. Впервые описан Н. А. Шерешевским в 1925 г., а в 1938 г.— 1. Turner. Встречается с частотой 1 : 2500—1 : 3000 новорожденных девочек.
Патоморфологические исследования указывают на недоразвитие половых желез, которые или вовсе отсутствуют, или имеют вид соединительнотканных тяжей с остатками яичниковой ткани и интерстициальных клеток. Нередко обнаруживают пороки развития сердечно-сосудистой системы (коарктацию аорты, стеноз легочной артерии, дефект межжелудочковой перегородки, незаращение боталлова протока), желудочно-кишечного тракта, мочевыделительной системы (кистозная почка, подковообразные почки).
Диагностировать синдром можно уже в период новорожденности. Дети рождаются с низкой массой и небольшим ростом, умеренная отечность кистей и стоп может наблюдаться в течение нескольких месяцев; низкий рост волос на шее, шея короткая с крыловидными складками, идущими от сосцевидных отростков к плечам, или избыточная подвижность кожи на шее. Из других аномалий развития можно отметить эпикант, сросшиеся брови, птоз, лагофтальм или экзофтальм, гипертелоризм, микрофтальм, колобомы век, широкую плоскую грудную клетку, имитирующую широко расставленные соски, сращение позвонков, клинодактилию, вальгусное искривление стоп, аномалии прикуса, телеангиэктазии кожи и кишечника, остеопороз.
При офтальмологическом исследовании выявляются облаковидные помутнения и снижение чувствительности роговицы, бледность зрительного нерва, сужение артерий на глазном дне, микрофтальм, катаракта.
В неврологическом статусе обычно отклонений не наблюдается, за исключением общей мышечной гипотонии. Психическое развитие в раннем возрасте нормальное или темп его несколько замедлен.
При дерматоглифическом исследовании выявляется изменение кожных узоров пальцев и ладоней. Обычно обнаруживают увеличение частоты ульнарных петель на больших и указательных пальцах. Дистальный осевой трирадиус встречается у 50% больных с синдромом Шерешевского—Тернера. Чаще, чем у здоровых, наблюдается поперечная складка ладони и единственная складка на V пальце. Ладонные узоры очень большие дистальные петли или завитки с большим гребневым счетом.
Комплекс указанных симптомов является показанием для исследования соскобов слизистой оболочки полости рта на половой хроматин. Около 80% больных с синдромом Шерешевского— Тернера являются хроматин-негативными, их кариотип 45, ХО. При делециях Xq-, Xp-, а также при кольцевой Х-хромосоме, мозапцизме ХО/ХХ клинические признаки менее выражены, чем при синдроме ХО. В соскобах со слизистой определяются мелкие тельца Барра в меньшем количестве, чем у нормальных девочек.
Диагноз верифицируется исследованием кариотипа лимфоцитов периферической крови.
В раннем возрасте синдром следует дифференцировать от гипотрофии другой этиологии, гипотиреоза, врожденных аномалий развития нехромосомной природы; при выраженной избыточности кожи на шее — от cutis laxa и синдрома Элерса — Данлоса.
Лечение синдрома Шерешевского—Тернера в раннем возрасте симптоматическое. С целью стимуляции психического и моторного развития применяют церебролизин, аминолон, ацефен, префизон, витамины группы В, массаж, лечебную физкультуру.
Синдром XXY (синдром Клайнфельтера)
Синдром обусловлен трисомией половых хромосом вследствие наличия добавочной Х-хромосомы. Встречается с частотой 1 : 400—500 новорожденных мальчиков. Описан в 1942 г. A. Klinefelter и др.
Патоморфологически характеризуется первичной дисгенезией гонад. При их гистологическом исследовании обнаруживают сужение или облитерацию семенных канальцев, гиалиновый склероз, разрастание клеток Лейдига. Необлитерированные канальцы заполнены дегенеративными сертолиевыми клетками.
Характерным признаком синдрома в раннем детском возрасте является уменьшение размера яичек и изменение их консистенции (более плотные или, наоборот, более мягкие). Уже в период новорожденности обращают на себя внимание особенности телосложения ребенка — непропорционально длинные ноги и руки, узкая грудная клетка. Психическое развитие чаще нормальное. У ряда больных описывают изменения глаз в виде пигментной дегенерации сетчатки и колобомы увеального тракта.
Мальчики с синдромом Клайнфельтера хроматин-положительны. На дерматоглифах может быть смещение осевого трирадиуса, увеличение угла atd, увеличение частоты дуг на пальцах, тенденция к уменьшению гребневого счета.
Диагноз верифицируется исследованием кариотипа в лимфоцитах периферической крови, при котором в большинстве случаев выявляется 47 хромосом вследствие добавочной Х-хромосомы. Однако число Х-хромосом может быть и больше 2. У таких больных все симптомы заболевания более выражены, обязательно отмечается задержка психического развития, тем более глубокая, чем больше Х-хромосом в кариотипе, могут быть гигантизм, акромегалия.
В раннем возрасте синдром диагностируют лишь при скринирующем исследовании полового хроматина.
Лечение в раннем возрасте проводят лишь в случаях, протекающих с отставанием в психическом развитии. Назначают препараты, стимулирующие функцию центральной нервной системы (аминолон, церебролизин, витамины группы В), проводят логопедические и педагогические занятия, целенаправленно формирующие высшие корковые функции.
Синдром XYY. Кариотип 47, XYY встречается среди новорожденных мальчиков с частотой 1 : 250—500 и чаще всего не сопровождается патологическим фенотипом. В раннем возрасте особенностей развития не обнаруживают. Может быть случайной кариолоптческой находкой.
Синдром полисомии X. Чаще всего встречается в виде трисомии X (47, XXX) и может не сопровождаться патологическим фенотипом. При числе Х-хромосом больше 3 характерны задержка психического развития и дисгенезация гонад тем более выраженные, чем больше добавочных Х-хромосом.
– Вернуться в оглавление раздела “Неврология.”
Оглавление темы “Наследственные синдромы в неврологии”:
1. Установление типа наследования в неврологии. Виды наследования неврологических заболеваний
2. Половой хроматин в неврологии. Дерматоглифика
3. Хромосомные синдромы в неврологии. Синдром трисомии 21-й хромосомы – синдром Дауна
4. Синдром трисомии 18-й хромосомы. Синдром Эдвардса в неврологии
5. Синдром Патау в неврологии. Синдром трисомии D2
6. Синдром Лежена. Синдром крик кошки в неврологии
7. Синдром Вольфа — Хиршорна. Синдром делеции длинного плеча хромосомы D
8. Синдром делеции 18-й хромосомы. Синдром кошачьих глаз – синдром Шмида—Фраккаро
9. Синдром делеции хромосом группы G. Синдромы дупликации-делеции с частичной трисомией 9-й хромосомы
10. Синдром Х0-синдром Шерешевского—Тернера. Синдром XXY – синдром Клайнфельтера в неврологии
Источник