
Сообщение на тему синдром марфана

Приветствую, дорогие читатели. Сегодня хотел бы поговорить про высоких людей с Синдромом Марфана, для которых высокий рост – это не просто особенность организма, а симптом серьезной болезни, требующей к себе постоянного внимания.
Синдром Марфана — заболевание наследственного типа, при котором поражается соединительная ткань с вовлечением в процесс скелетно-мышечной системы и глаз. Установлено, что причиной патологии является мутация гена фибриллина FBN1. Заболевание полиморфно — может протекать с разной выраженностью клинической картины, и характеризуется появлением все новых типов мутации в генах.
Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил описание 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.
Распространенность синдрома — 1 случай на 10000 человек. Риск рождения ребенка с синдромом Марфана повышается после достижения отцом возраста 35 лет и достигает 50% при наличии патологии у одного из родителей. Врожденная аномалия наследуется по аутосомно-доминантному типу. В ее основе лежит дефект важнейшего гена, отвечающего за синтез коллагена.

Во время внутриутробного развития происходит нарушение формирования волокон соединительной ткани, утеря ими прочности, в результате чего волокна не способны выдерживать естественные нагрузки. Поэтому наибольшие атипичные изменения претерпевают крупные сосуды, клапаны сердца, связки глаза, твердое небо, скелет и мышцы.
Без адекватной терапии продолжительность жизни людей с синдромом Марфана не более 40 лет. Терапия позволяет увеличить этот срок вдвое и более.
История заболевания
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.
Симптомы синдрома Марфана
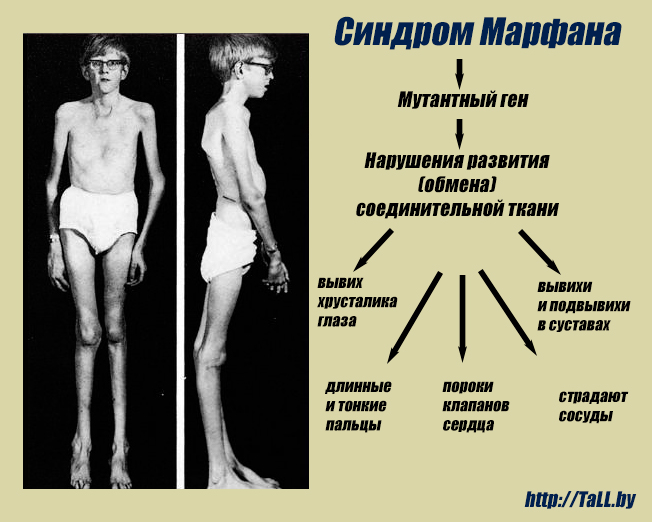
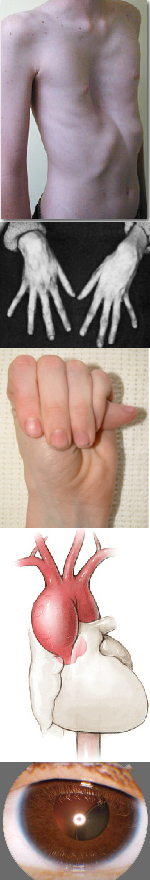
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия) Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
 Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
— гиперподвижность суставов;
— аномалии строения тазобедренного сустава;
— кифоз, сколиоз;
— вывихи шейного сегмента позвоночника;
— деформация грудной клетки;
— плоскостопие;
— глубокая посадка глаз;
— уменьшенная нижняя челюсть, нарушение роста зубов;
— высокое нёбо;
— атрофические «растяжки» на коже;
— паховые грыжи, частые разрывы связок.
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);
— пороки сердца (чаще — поражения клапанов);
— стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия). Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности. Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
Лечение и профилактика осложнений
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана. Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.

Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения:
— прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.);
— хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
Известные люди с синдромом Марфана
Хоть синдром Марфана – очень редкое заболевание, есть немало знаменитостей, больных синдромом Марфан: Фло Хайман (призер Олимпийских игр по волейболу), Джон Тавенер (композитор), Джоуи Рамон (музыкант), Лесли Хорнби (фотомодель и певица) и другие.
Среди исторических личностей, известных во всем мире, с синдромом Марфана можно выделить:

Музыкант-скрипач Никколо Паганини. Поскольку Паганини умер еще до описания синдрома, данные о его заболевании исследуются по сохранившимся изображениям и дневнику лечащего врача. Скрипач имел характерную деформацию пальцев, высокий рост и худобу, непропорциональное развитие конечностей, впалую грудь, мышечную слабость.
Писатель Ганс Кристиан Андерсен. Имел угловатое лицо, был очень худым и длинноруким, рано заполучил проблемы со зрением.
Президент Америки Авраам Линкольн. Кроме всех внешних признаков синдрома Марфана у Линкольна наблюдались ревматические боли, «разболтанность» суставов, но, в то же время — хорошая физическая выносливость.
Писатель Корней Чуковский. Наличие большого непропорционального носа, длинных конечностей не помешало Чуковскому стать одним из лучших творцов современности и доктором филологии.
Осама Бен Ладен. «Террорист №1» мира имел высокий рост и малый вес и большие проблемы с суставами и позвоночником, а также вытянутый череп и слишком узкое лицо.
Источники и ссылки:
1. Причины высокого роста — статья, в которой рассмотрены основные причины высокого роста человека.
2. Синдром Марфана в википедии — о синдроме Марфана в свободной энциклопедии.
3. Русскоговорящее сообщество людей с синдромом Марфана — форум о синдроме Марфана, общение, обсуждение болезни.
4. Международный фонд синдрома Марфана — фонд помощи больным с синдромом Марфана.
5. Группа вконтакте для больных синдромом Марфана.
Источник
Синдром Марфана – дифференцированная форма врожденной соединительнотканной недостаточности, характеризующаяся разнообразными проявлениями скелетной, сердечно-сосудистой и глазной патологии. У больных с синдромом Марфана отмечаются гигантизм, долихостеномелия и арахнодактилия, аневризмы аорты, миопия, эктопия хрусталика, деформация грудины, кифосколиоз, плоскостопие, протрузия вертлужной впадины, эктазия твердой мозговой оболочки. Диагноз синдрома Марфана основан на семейном анамнезе, результатах функционального, офтальмологического, рентгенологического и генетического исследований. Лечение при синдроме Марфана включает консервативную и хирургическую коррекцию сердечно-сосудистых нарушений, поражений скелета и органа зрения.
Общие сведения
Синдром Марфана – системное недоразвитие соединительной ткани в эмбриональном и постнатальном периодах, обусловленное структурными дефектами коллагена и сопровождающееся преимущественным поражением опорно-двигательного аппарата, глаз, сердечно-сосудистой системы. Синдром Марфана – одна из наиболее распространенных наследственных коллагенопатий синдромального характера. Частота встречаемости синдрома Марфана в популяции невысока: по данным различных авторов составляет 1 случай на 10000-20000 человек, без расовой и половой детерминированности.

Синдром Марфана
Причины синдрома Марфана
Синдром Марфана относится к врожденным аномалиям, наследуемым по аутосомно-доминантному типу, с выраженным плейотропизмом, варьирующей экспрессивностью и высокой пенетрантностью. В основе синдрома Марфана лежат мутации в гене FBN1, отвечающем за синтез фибриллина – важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. Аномалия и дефицит фибриллина при синдроме Марфана приводят к нарушению формирования волокнистых структур, потере прочности и упругости соединительной ткани, невозможности выдерживать физиологические нагрузки. Гистологическим изменениям в большей степени подвержены стенки сосудов эластического типа и связочный аппарат (в первую очередь, аорта и цинновая связка глаза, содержащие наибольшее количество фибриллина).
Широкий фенотипический спектр синдрома Марфана (от легких форм, трудно отличимых от нормы до тяжелых, быстропрогрессирующих) объясняется разнообразием мутаций в гене FBN1 (более 1000 видов), а также присутствием мутаций в других генах (например, в гене трансформирующего фактора роста – TGFBR-2). При генетическом исследовании в 75% случаев синдрома Марфана выявляется семейный тип наследования, в остальных – первичная мутация. Риск рождения ребенка с синдромом Марфана возрастает с увеличением возраста отца (особенно после 35 лет).
Классификация синдрома Марфана
В зависимости от количества пораженных систем выделяют несколько форм синдрома Марфана:
- стертую – со слабо выраженными изменениями в 1-2-х системах
- выраженную – со слабо выраженными изменениями в 3-х системах; выраженными изменениями хотя бы в 1-ой системе; выраженными изменениями в 2-3-х и более системах.
Степень тяжести изменений при синдроме Марфана может быть легкой, средней и тяжелой. По характеру течения дифференцируют прогрессирующий и стабильный синдром Марфана.
Симптомы синдрома Марфана
Синдром Марфана характеризуется сочетанным поражением скелета, глаз, сердечно-сосудистой и нервной систем; многообразием проявлений, варьированием сроков появления первых признаков заболевания; хроническим прогредиентным течением.
Больные синдромом Марфана, как правило, отличаются высоким ростом, относительно коротким туловищем с непропорционально длинными тонкими конечностями (долихостеномелией) и удлиненными паукообразными пальцами (арахнодактилией); астеническим телосложением со слаборазвитой подкожной клетчаткой и мышечной гипотонией; длинным и узким лицевым скелетом (долихоцефалией); наличием высокого аркообразного неба и нарушения прикуса (прогнатии). Средняя длина тела при рождении у мальчиков с синдромом Марфана составляет 53 см, окончательный рост – 191 см; у девочек – соответственно 52,5 см и 175 см.
При синдроме Марфана отмечаются нарушение функции суставов (гипермобильность); деформация грудной клетки (воронкообразная или килевидная форма), деформация позвоночника (сколиоз, кифоз, кифосколиоз, подвывихи и вывихи шейного отдела, спондилолистез), а также плоскостопие и протрузия вертлужной впадины.
Сердечно-сосудистая патология, доминирующая в клинической картине синдрома Марфана и часто определяющая его исход, проявляется дефектами структуры стенок сосудов эластического типа, особенно аорты и крупных ветвей легочной артерии, пороками развития клапанного аппарата и перегородок сердца. Изменения аорты у больных синдромом Марфана характеризуются прогрессирующим расширением ее восходящей части и клапанного кольца (дилатацией, аннулоаортальной эктазией) и аневризмами; поражение митрального клапана – миксоматозной дегенерацией створок, патологическим удлинением и разрывом створочных хорд, обызвествлением клапанного кольца. У плода с синдромом Марфана возможно формирование врожденных пороков сердца – коарктации аорты, стеноза легочной артерии, ДМПП и ДМЖП. Органические и функциональные изменения сердца и сосудов у больных синдромом Марфана часто сопровождаются нарушением ритма (наджелудочковой и желудочковой тахикардией, фибрилляцией предсердий) и развитием инфекционного эндокардита.
Самая неблагоприятная неонатальная форма синдрома Марфана проявляется в классическом варианте уже при рождении, приводит к прогрессирующей сердечной недостаточности и летальному исходу на первом году жизни ребенка.
Для большинства случаев синдрома Марфана характерна патология органа зрения, включающая близорукость, вывих/подвывих (эктопию) хрусталика, уплощение и увеличение размера роговицы, гипоплазию радужной оболочки и цилиарной мышцы, косоглазие, изменение калибра сосудов сетчатки. Эктопия хрусталика при синдроме Марфана имеет двухсторонний характер, часто развивается в возрасте до 4-х лет и устойчиво прогрессирует, ухудшая зрительную функцию.
При синдроме Марфана наблюдается поражение других систем и органов: нервной (эктазия твердой мозговой оболочки, в т. ч. пояснично-крестцовое менингоцеле), бронхолегочной (спонтанный пневмоторакс, эмфизема легких, дыхательная недостаточность), кожи и мягких тканей (атрофические стрии), рецидивирующие паховые и бедренные грыжи, вывихи и разрывы связок, а также эктопия почек, опущение мочевого пузыря и матки, варикозное расширение вен и др.
Характерный для синдрома Марфана высокий выброс адреналина может способствовать постоянному нервному возбуждению, гиперактивности, а иногда развитию неординарных способностей и умственной одаренности.
Диагностика синдрома Марфана
Диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
За диагностические критерии синдрома Марфана берутся характерные изменения в различных системах и органах; главными (большими) из них считаются: дилатация корня/расслоение восходящей части аорты, эктопия хрусталика и эктазия твердой мозговой оболочки; килевидная/воронкообразная деформация грудной клетки, требующая хирургического лечения; отношение длины верхнего сегмента тела к нижнему < 0,86 или размаха рук к росту > 1,05; сколиоз (> 20˚) или спондилолистез; ограничение разгибания в локтевом суставе (<170о); плоскостопие; протрузия вертлужной впадины. Остальные проявления относятся к малым критериям, а генетические (семейные) признаки – к дополнительным. Для установления диагноза синдрома Марфана необходимо наличие минимум по 1-му большому критерию в двух системах органов и 1-го малого – в третьей; в скелетной системе – присутствие минимум 4-х больших.
Также применяются фенотипические диагностические тесты, определяющие соотношение кисть/рост (при синдроме Марфана > 11%); длину среднего пальца (> 10 см); индекс телосложения Варги – (масса тела, г/(рост, см)x2 – возраст, годы/100, должно быть <1,5); тест большого пальца на арахнодактилию, тест охвата запястья.
ЭКГ при синдроме Марфана позволяет определить нарушение ритма сердца, выраженную гипертрофию миокарда; ЭхоКГ – обнаружить клапанную регургитацию, увеличение размеров левого желудочка, пролапс митрального клапана, разрывы хорд, дилатацию аорты. На рентгенографии грудной клетки можно увидеть расширение корня и дуги аорты, увеличение размеров сердца; на КТ и МРТ сердца и сосудов – выявить дилатацию и аневризмы аорты.
Аортография показана при подозрении на аневризму и расслоение аорты. Наличие эктопии хрусталика уточняют с помощью биомикроскопии и офтальмоскопии; протрузию вертлужной впадины устанавливают методом рентгенографии тазобедренных суставов; эктазию твердой мозговой оболочки – МРТ позвоночника.
При синдроме Марфана определяется возрастание (в 2 раза и более) почечной экскреции метаболитов соединительной ткани: глюкозоаминогликанов и их фракций. Метод прямого автоматического секвенирования ДНК позволяет провести генетическую идентификацию мутаций в гене FBN1.
Необходима дифференциальная диагностика с заболеваниями, внешне напоминающими синдром Марфана: гомоцистинурией, врожденной контрактурной арахнодактилией (синдромом Билса), наследственной артроофтальмопатией (синдромом Стиклера), MASS-синдромом, синдромами Элерса-Данлоса, Лойса-Дитца, Шпринцена–Голдберга, семейной эктопией хрусталика и др.
Лечение синдрома Марфана
Лечение и дальнейшее наблюдение пациентов с синдромом Марфана должно осуществляться группой специалистов: офтальмологом, кардиологом, кардиохирургом, ортопедом, генетиком, терапевтом.
Лечение больных с синдромом Марфана направлено на профилактику прогрессирования заболевания и развития осложнений, в первую очередь в сердечно-сосудистой системе. При диаметре аорты до 4 см назначаются β-адреноблокаторы, антагонисты кальция или ингибиторы АПФ. Хирургическое лечение проводится при недостаточности клапанов сердца, пролапсе митрального клапана, значительном расширении (>5 см) восходящей части и расслоении аорты. Реконструктивные операции на аорте при синдроме Марфана, имеют высокий процент послеоперационной 5-ти и 10-ти летней выживаемости. При необходимости выполняют протезирование митрального клапана. У беременных с синдромом Марфана и выраженной сердечно-сосудистой патологией проводят досрочное оперативное родоразрешение путем кесарева сечения. С целью профилактика инфекционного эндокардита и тромбозов после операционных вмешательств назначаются антибиотики и антикоагулянты.
При синдроме Марфана проводится коррекция зрения с помощью подбора очков и контактных линз, при необходимости – лазерное или хирургическое лечение катаракты, глаукомы, удаление смещенного хрусталика с имплантацией искусственного. При выраженных скелетных нарушениях может потребоваться хирургическая стабилизация позвоночника, торакопластика, эндопротезирование тазобедренных суставов. Применяются также патогенетическая коллагеннормализующая терапия, метаболическая и витаминотерапия.
Прогноз
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечно-сосудистых изменений, а также поражений скелета и глаз. Имеется высокий риск осложненного течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти. Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование. При синдроме Марфана показан низкий или средний уровень физической активности, исключающий занятия контактными видами спорта, спортивные соревнования, изометрические нагрузки, подводное плавание. Женщинам детородного возраста с синдромом Марфана необходимо пройти медико-генетическое консультирование.
Источник