
У человека синдром марфана наследуется по принципу

Синдром Марфана — генетическая патология, при которой нарушен синтез фибриллина в организме. Это белок, отвечающий за эластичность и сократимость соединительной ткани. При данном заболевании поражаются кости скелета, развиваются сердечно-сосудистые патологии, а также возникают нарушения зрения. Читайте об этом подробнее в статье.
Синдром Марфана диагностируется с частотой один случай на 5 тысяч человек (в любой этнической группе). Если не проводить лечебные мероприятия, то продолжительность жизни таких больных невысока — примерно 30-40 лет. В странах с высоким уровнем здравоохранения люди с таким синдромом успешно лечатся и доживают до преклонного возраста.
Синдром Марфана — что это? Тип наследования

Данное заболевание имеет аутосомно-доминантный характер. В его основе лежат мутации в гене FBN1, отвечающем за синтез фибриллина — одного из самых важных белков для нормальной жизнедеятельности, который поддерживает эластичность соединительной ткани. При болезни Марфана возникает его недостаток, а это приводит к тому, что соединительная ткань теряет прочность и упругость, человек не может выдерживать большие физиологические нагрузки. Также страдает аорта и цинновая связка глаза, которая содержит большое количество фибриллина. Эластических волокон много во всем теле человека, но самое большое их сосредоточение именно в этих структурах. Нередко смерть больного происходит именно от аневризмы аорты.
Медицинские исследования подтверждают, что в 75% случаев синдром Марфана передается по семейному признаку, и лишь в остальных 25% случаев это обусловлено первичной спорадической мутацией.
Также было выявлено, что вероятность рождения младенца с синдромом Марфана увеличивается в зависимости от возраста отца: чем он старше, тем больше риск.
Проявления заболевания
Данной патологии сопутствует множество нарушений в организме. Так называемая «триада Марфана» — это повреждение скелетных костей, болезни сердечно-сосудистой системы и различные нарушения зрения. Однако надо отметить, что на умственные способности этот синдром никак не влияет.
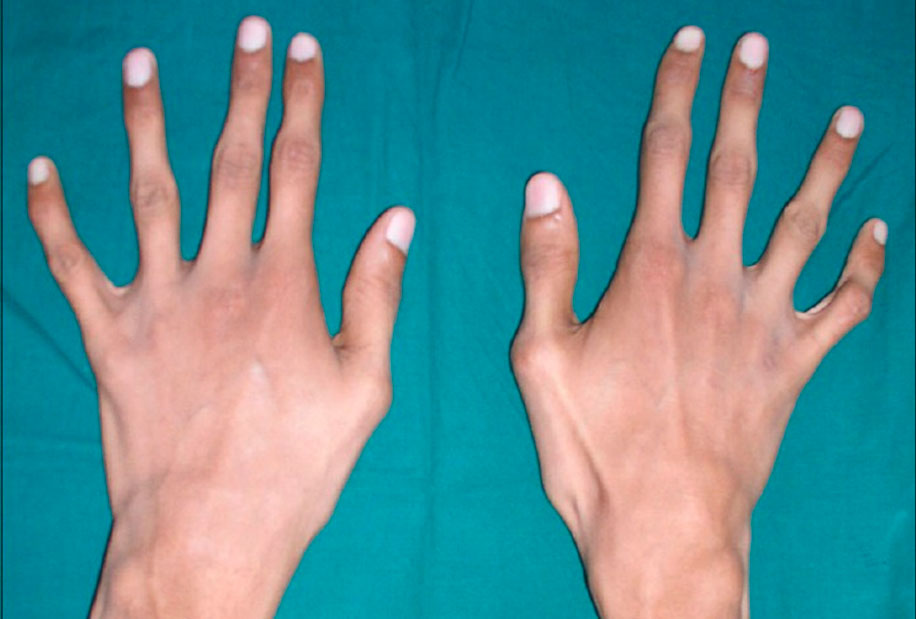
Заболевшие отличаются, как правило, непропорциональным сложением: при довольно высоком росте у них укороченное туловище, длинные руки и пальцы, напоминающие паучьи (арахнодактилия), астеническое телосложение с мышечной гипотонией, деформация грудной клетки, сильное искривление позвоночника (кифоз, сколиоз, подвывихи и вывихи шейного отдела), плоскостопие.
Синдром Марфана, сопровождающийся аномалией сердечно-сосудистой системы, выражается в дефектах стенок крупных сосудов, особенно аорты и больших ветвей легочных сосудов, пороками развития сердца. Эти изменения часто формируются уже у развивающегося плода. Самая опасная форма синдрома Марфана, симптомы которой проявляются уже при рождении, приводит к прогрессирующей сердечной недостаточности и летальному исходу на первом году жизни младенца.
Синдром Марфана у детей и взрослых, сопровождающийся аномалией развития зрительного аппарата
Различные поражения органов зрения при данной болезни диагностируются у 50-80% пациентов, причем нарушения начинают проявляться уже в детском возрасте. Это могут быть следующие патологии:
- эктопия хрусталика;
- сильная близорукость;
- уплощение и увеличение размера роговицы;
- косоглазие;
- гипоплазия радужной оболочки и цилиарной мышцы;
- изменения в сетчатке;
- деструкция стекловидного тела и другие.
Терапия глазных заболеваний проводится обычными в таких случаях методами. Однако их воздействие может быть затруднено текущими процессами в организме. Рассмотрим подробнее офтальмологические патологии, сопутствующие синдрому Марфана.
Близорукость
Данное нарушение зрения проявляется более чем у половины больных. Причинами ее могут быть: шаровидная форма хрусталика, изменение преломляющей силы роговицы глаза вследствие ее уплощения и увеличения, а также быстрый рост по переднезадней оси самого глазного яблока. При этом миопия может достигать довольно высоких степеней.
В детском возрасте патология лечится с помощью очков. Начиная с 8-10 лет, можно также использовать контактные линзы.
У больных детей также нередко может развиваться синдром «ленивого глаза» — амблиопия. Родителям маленьких пациентов с синдромом Марфана следует быть очень внимательными к состоянию зрения ребенка. Амблиопия зачастую переходит в косоглазие или наоборот.
Эктопия хрусталика
Эктопия хрусталика глаза — его смещение со своего обычного положения. Из-за растяжения и слабости цинновых связок, на которых он держится, происходит их разрыв. Если повреждение частичное, то хрусталик смещается, но все-таки держится на оставшихся связках — это называется подвывихом. Если же связки Цинна отрываются полностью, то он опускается в полость глаза, свободно меняя свое положение — такое состояние называется вывих.
При синдроме Марфана цинновы связки слабы или недоразвиты, поэтому полная или частичная эктопия хрусталика — нередкое явление.
Основным симптомом этого зрительного нарушения является дрожание радужки — иридодонез. Глубина передней камеры глаза в стороне смещения хрусталика становится более мелкой. Обнаружить иридодонез может врач путем инструментального исследования. Больной со своей стороны испытывает диплопию — двоение в глазах и ухудшение четкости видения. Если вывих произошел в переднюю камеру зрительного органа, то его можно заметить извне — он похож на золотистую каплю масла. При этом в области глаз может отмечаться боль и гиперемия.
Частичный неосложненный подвывих хрусталика не требует специальной терапии. Для поддержания качества зрения назначаются подходящие контактные линзы. Иногда для его устранения используют способ малоинвазивной транссклеральной фиксации ИОЛ. В поврежденный глаз имплантируют специальное устройство, выполняющее функцию цинновых связок — оно прочно удерживает хрусталик на его анатомическом месте.
Катаракта и глаукома при синдроме Марфана
Осложнения при эктопии хрусталика могут проявиться в его помутнении — тогда развивается катаракта. При этом качество зрения постепенно падает, и процесс необратим. Лечить заболевание надо своевременно. Обычно операция назначается при помутнении примерно 50% хрусталика. Современные методы в офтальмологии позволяют без труда справиться с этим нарушением зрения. Для этого проводится хирургическая операция: хрусталик удаляется посредством факоэмульсификации, а вместо него имплантируется интраокулярная линза. Она заменяет естественную и обеспечивает высокое качество зрения надолго.
При синдроме Марфана также может развиться вторичная глаукома — состояние, при котором блокируется поступление внутриглазной жидкости из задней камеры глаза в переднюю, в связи с чем резко повышается внутриглазное давление.
В данной ситуации глаукома лечится теми же способами, что и обычно. Назначаются средства, понижающие давление и увеличивающие отток внутриглазной жидкости. В более тяжелых случаях может быть назначена хирургическая операция, направленная на улучшение оттока жидкости.

Болезни радужной оболочки
Синдрому Марфана нередко сопутствуют изменения в радужной оболочке — это связано с повышенной растяжимостью тканей. Могут возникнуть колобомы, гипоплазия или атрофия радужки с нарушением ее диафрагмальной функции. Колобома — это дефект, проявляющийся в отсутствии части глазной оболочки. Обычно имеет грушевидную форму и располагается в нижней части радужки. К наиболее ранним проявлениям у людей с синдромом Марфана относят гипоплазию стромы радужки, особенно ее пигментной зрачковой каймы. Слабость дилататора (мышцы-расширителя) у больных не позволяет достичь полного расширения зрачка даже с помощью мидриатиков.
Патологии сетчатки при синдроме Марфана
Из-за слабости соединительной ткани подвержена растяжению также сетчатка глаза. В результате этого повышается риск появления периферических хориоретинальных дистрофий — локальных истончений сетчатки, которые могут спровоцировать отторжение слоя светочувствительных клеток от пигментного эпителия. Такое нарушение очень опасно: начинает падать острота зрения, ухудшается восприятие света и цвета.
Признаками отслойки сетчатки могут быть следующие проявления:
- вспышки, искры в глазах — такое явление называется фотопсией;
- искажение формы, размера и оттенка объектов — метаморфопсия;
- «мушки» и черные точки перед глазами как следствие повреждения ретинального сосуда;
- выпадение из поля зрения отдельных элементов в видимой картинке — это признак того, что отслоение началось в центральной зоне сетчатки;
- появление темной пелены, охватывающей все большую область, снижение периферийной видимости.
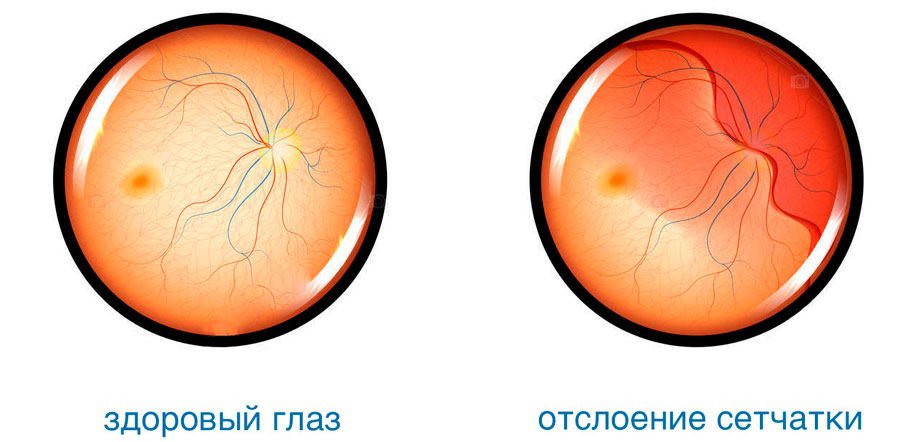
Отслоение сетчатки сегодня поддается успешному лечению различными методами. Наиболее эффективным из них является лазерная коагуляция — прижигание поврежденных участков с целью надежного их соединения с сосудистой оболочкой.
Как ставится диагноз «синдром Марфана»?
Нет никаких специальных тестов, которые бы позволили установить наличие этой патологии. Для его подтверждения нужно пройти обследование у нескольких специалистов. При этом симптомы болезни могут обнаружиться не сразу после рождения, а в течение жизни ребенка — иногда это происходит и в 10, и в 16 лет.
Ортопед выявит нарушения, связанные с костными структурами: искривление позвоночника и деформацию грудной клетки, а также плоскостопие, удлиненность трубчатых костей скелета, гипермобильность суставов.
Специалист по генетике соберет семейный анамнез, чтобы выявить родственников, умерших от сердечно-сосудистых заболеваний.
Офтальмолог проведет обследование с помощью щелевой лампы, а также используя другие современные методы проверки органов зрения, чтобы выявить имеющиеся глазные патологии.

Кардиолог назначит рентген грудной клетки, электрокардиограмму, эхокардиограмму и другие методы. Обычно после сбора сведений у всех специалистов назначается небольшой консилиум, где врачи ставят окончательный диагноз. В этом случае важно принять точное решение. Не все люди с длинными и тонкими пальцами, искривлением позвоночника имеют синдром Марфана. С другой стороны, подобными симптомами могут сопровождаться другие болезни, связанные со слабостью соединительной ткани.
Контроль и лечение заболевания
Дети с синдромом Марфана должны постоянно быть под медицинским и домашним наблюдением. Их организм растет и развивается довольно быстро, особенно в подростковый период, и тогда нужны будут осмотры окулиста, ортопеда и других специалистов, проведение при необходимости лечебных мероприятий. Если соблюдать рекомендации для больных синдромом Марфана, то можно сохранить здоровье надолго.
Очень важно не подвергать сердце высокой нагрузке, учитывая вероятность разрыва аорты или крупных сосудов в любой момент. И детям, и взрослым противопоказаны виды спорта, предполагающие бег, сильное мышечное напряжение или риск получить удар в грудную клетку: баскетбол, волейбол, бокс, тяжелая атлетика и подобные спортивные дисциплины.
Это не означает, что активность им противопоказана совсем. Такие люди могут и должны поддерживать физическую форму. Например, езда на велосипеде, медленные танцы, пешие прогулки, плавание вполне подходят в данной ситуации.

Многие проявления синдрома Марфана контролируются с помощью лекарств, а при необходимости — хирургическими способами. Так, бета-адреноблокаторы применяют для понижения артериального давления и уменьшения износа стенок сосудов, что тормозит процесс прогрессирования аневризмы аорты. Если же она достигает слишком больших размеров или беспокойство вызывает сердечный клапан, может быть назначена хирургическая операция.
Детям, страдающим миопией или синдромом «ленивого глаза», назначаются коррекционные очки или контактные линзы.
При развитии сколиоза, возможно, придется носить специальный поддерживающий корсет, заниматься ЛФК. В особо тяжелых ситуациях при сильной деформации грудной клетки или позвоночника может потребоваться операция. В некоторых странах дети и подростки с синдромом Марфана носят браслет с информацией. Это понадобится в критическом случае, чтобы врачи знали диагноз пациента.
Советы родителям ребенка с синдромом Марфана
Конечно, грустно и горько осознавать, что у ребенка имеется такое опасное заболевание. Однако только правильное поведение родителей поможет преодолеть трудности, в том числе создать нужный психологический настрой. Нужно научиться жить с этой проблемой и постоянно держать под контролем состояние ребенка. Важно найти хороших специалистов, которые будут заниматься его здоровьем.
Следует также научить его относиться с достоинством к своему состоянию, не реагировать на возможные насмешки со стороны. Лучше привить ему любовь к занятиям, которыми он сможет заниматься в дальнейшем: пусть это будет программирование или, например, музыка. Если диагноз был поставлен уже в старшем возрасте, то придется отказаться от некоторых видов спорта.

Важно также наладить хороший контакт с учителями, объяснив им, что это заболевание предполагает некоторые особенности. Например, сидеть ему лучше за первой партой из-за проблем со зрением, не стоит испытывать слишком сильных физических нагрузок. Каждому родителю хочется обеспечить своему ребенку счастливое детство. Дети с синдромом Марфана должны знать, что в мире много интересных занятий, которые им доступны.
Болезнь не поддается какому-либо конкретному лечению. К сожалению, медики пока не научились изменять мутированные гены. Однако врачи подскажут целый ряд вариаций терапии, направленной на улучшение состояния конкретного органа и предотвращения развития осложнений.
Источник
В последние годы отмечается значительный рост наследственных заболеваний с поражением соединительной ткани. Некоторые специалисты считают, что причина такого явления кроется в накоплении в процессе эволюции человека новых генетических изменений (мутаций), к чему приводят внутренние и внешние факторы окружающей среды. Другие полагают, что на самом деле просто возросли диагностические возможности в связи с совершенствованием медико-генетических знаний, которые позволяют распознать генетическую патологию и поставить правильный диагноз.
Большую группу наследственных заболеваний составляет поражение соединительной ткани. Так как она является неотъемлемой частью всех органов и систем в организме, то такие патологии отличаются множеством нарушений и клинических симптомов. Одним из самых известных генетических заболеваний с поражением соединительной ткани считается синдром Марфана.
Такая патология характеризируется большой вариабельностью симптоматики (от скрытых форм до несовместимых с жизнью вариантов течения) и чаще всего включает поражения сердечно-сосудистой системы, глаз, центральной нервной системы и опорно-двигательного аппарата.
Встречается заболевание достаточно редко – 1 случай на 10 000 человек. Но если обратиться к статистике европейских стран, то частота значительно выше – 1-3 случая на 5000 человек, что связано с большей доступностью специфической диагностики скрытых форм патологии.

Известные люди тоже болели синдромом Марфана
Причины и генетика патологии
Впервые болезнь детально описал в 1896 году французский педиатр А.Марфан, в честь которого и назвали патологию. Он наблюдал за 5-летней девочкой астенического телосложения с непропорционально длинными конечностями и врожденной арахнодактилией (длинными пальцами). К середине 20-х годов прошлого столетия уже имелось множество подобных описанных клинических случаев у детей и взрослых. Американский генетик Мак Кьюсик провел детальное исследование мутаций хромосом и открыл новую группу наследственных заболеваний соединительной ткани, куда и отнесли синдром Марфана.
По этиологии синдром Марфана является генетическим заболеванием с аутосомно-доминантным типом наследования, с различной степенью экспрессивности (клинических проявлений генетических изменений). Примерно 85% случаев недуга носит наследственный характер, то есть наследуется от родителей. Остальные случаи являются новыми, то есть возникают вследствие новых спонтанных мутаций, а не передаются по наследству.
Непосредственная причина патологии в 95% случаев – это мутация в гене, которые кодирует строение фибриллина-1 и/или фибриллина-2. Локализируется мутация гена FBN1 и FBN2 в хромосоме 15 и 3.

Синдром Марфана наследуется по аутосомно-доминантному типу
Фибриллин – это основа эластических волокон соединительной ткани гликопротеиновой природы. Он составляет каркас межклеточного вещества, сосудистых стенок, хрящей, хрусталика глаза и многих других органов и тканей. В случае наличия описанной мутации у пациента соединительная ткань отличается повышенной способностью к растяжению, становится менее прочной и выносливой к механическим воздействиям, что и становится причиной клинических проявлений синдрома.
Примерно в 5% случаев непосредственной причиной синдрома Марфана (атипичные формы патологии) является точковая мутация гена, который кодирует строение α2-цепи коллагена первого типа.
Классификация
Согласно МКБ-10, синдром Марфана входит в класс врожденных дефектов развития и хромосомных патологий, тут патологию можно найти под шифром Q87.4.
Детальной клинической классификации недуга на сегодняшний день не существует, но выделяют несколько форм болезни, в зависимости от тех или иных критериев.
В зависимости от выраженности симптомов:
- стертая форма – признаки патологии мало выражены и могут оставаться незамеченными на протяжении всей жизни, как правило, изменения касаются не более 2 систем органов;
- клинически выраженная форма – симптомы патологии хорошо заметны и встречаются более чем в 2 системах органов.

Внешний вид ребенка, больного синдромом Марфана
В зависимости от генетического фактора:
- семейная форма диагностируется в случаях, когда болезнь передается по наследству;
- спорадическая форма определяется тогда, когда патология обусловлена новой спонтанной мутацией у индивида и при этом не встречается у его родственников.
Симптомы синдрома Марфана
Признаки синдрома Марфана очень разнообразны. Поэтому их рассматривают с точки зрения поражения отдельных органов и систем:
- опорно-двигательного аппарата;
- органов зрения;
- сердечно-сосудистой системы;
- нервной системы;
- дыхательной системы;
- кожных покровов и мягких тканей;
- прочих органов и систем.
Опорно-двигательный аппарат
Патологическая соединительная ткань обусловливает развитие ряда специфических фенотипических признаков и деформаций скелета у пациентов с данной патологией.

Характерный внешний вид пациентов с синдромом Марфана
Как правило, внешне пациенты с синдромом Марфана выглядят достаточно специфически. Для них характерно:
- астеническое телосложение;
- высокий рост;
- плохое развитие подкожной жировой клетчатки, из-за чего люди выглядят худощавыми;
- очень длинные верхние и нижние конечности при относительно коротком туловище;
- череп вытянутый (долихоцефалический);
- удлиненные пальцы – паукообразные (арахнодактилия);
- лицо узкое, вытянутое по вертикали;
- готическое верхнее небо;
- недоразвитие скул;
- выступающая нижняя челюсть (прогнатизм);
- неправильный рост (скученность) зубов и патологический прикус;
- гипермобильность суставов, их «разболтанность;
- глубоко посажены в черепе глаза.

Пример того, как определить наличие арахнодактилии, характерной для синдрома Марфана
По мере роста ребенка могут появляться различные деформации скелета. Чаще всего появляются искривления позвоночного столба. У пациентов диагностируют сколиоз, патологический кифоз и лордоз, осанку по типу «прямой спины» (сглаживание физиологического поясничного лордоза). Также появляются подвывихи и вывихи в шейном отделе позвоночника, примерно у 20% больных диагностируют поясничный спондилолистез.

Выраженная степень арахнодактилии при синдроме Марфана со специфическими контрактурами пальцев
Среди других скелетных деформаций встречается:
- протрузия вертлужной впадины тазобедренного сустава 2-3 степени, которая становится причиной диспластического коксартроза и инвалидности многих пациентов, если не выполнить операцию эндопротезирования;
- килевидная и вдавленная деформация грудной клетки;
- плоскостопие (продольное и поперечное).
Для пациентов в СМ также характерна остеопения (снижение минеральной плотности костей) и частые патологические переломы костей на ее фоне, а также склонность к привычным вывихам, например, плеча.

Вдавленная деформация грудной клетки у пациента с синдромом Марфана и результат ее хирургической коррекции
Сердечно-сосудистая система
Среди поражений кардиоваскулярной системы при СМ чаще всего встречаются:
- пролапс створок митрального клапана с регургитацией или без
- миксаматоз сердца;
- дилятационная кардиомиопатия с развитием сердечной недостаточности;
- аневризмы аорты и других сосудов (мозговых, почечных, пр.);
- расширение легочной артерии и различных отделов аорты.
Именно кардиоваскулярные патологические изменения при СМ определяют прогноз и продолжительность жизни пациентов. Примерно 90% всех пациентов с данной генетической патологией умирают в возрасте 40-50 лет вследствие таких осложнений, как расслоение и разрыв аневризмы аорты, других сосудов, прогрессирующей недостаточности сердца вследствие дилатации его камер и изменений клапанного аппарата.
При наличии СМ возможно наличие врожденных пороков сердца у детей. Чаще всего встречаются коарктация аорты, стеноз (сужение) легочной артерии, дефект межжелудочковой и межпредсердной перегородки.
Также такие пациенты склонны к различным сердечным аритмиям, среди которых и опасные для жизни (мерцательная аритмия, желудочковая тахикардия и экстрасистолия), к инфекционному эндокардиту.
Аневризмы аорты и их разрыв чаще всего становятся причиной смерти пациента с синдромом Марфана
Орган зрения
Патологические изменения глаз являются весьма характерными для данного недуга. Примерно у 60-80% пациентов диагностируется дислокация хрусталика из-за слабости его связочного аппарата, причем еще в младенческом возрасте. Среди других характерных признаков:
- уплощение роговицы;
- увеличение размеров глазного яблока в длину;
- миопия или гиперметропия;
- нарушение процесса аккомодации из-за недоразвития цилиарной мышцы.
В случае выявления описанных поражений органа зрения у новорожденного ребенка следует задуматься о возможной генетической патологии.

Дислокация хрусталика – типичный признак синдрома Марфана
Нервная система
Из-за патологического строения стенок сосудов у пациентов с СМ повышен риск геморрагических инсультов, а также кровоизлияний в мозг при разрыве сосудистых аневризм, субарахноидальных кровотечений.
Среди аномалий развития встречается эктазия твердой мозговой оболочки. Чаще всего приходится сталкиваться с пояснично-крестцовой эктазией мозговой оболочки (выпячивание твердой мозговой оболочки за пределы позвоночного канала через дефект в строении позвонков). Это большой критерий СМ, который встречается в 40% случаев заболевания.
У части пациентов встречаются отклонения в интеллектуальном развитии, но большинство людей с СМ характеризируются высокими показателями IQ.
Органы системы дыхания
В большинстве случаев изменения бронхолегочного аппарата диагностируются случайно. Характерно развитие булл в верхних частях легких, которые иногда могут разрываться с развитием спонтанного пневмоторакса.
Также из-за деформаций грудной клетки пациенты склонны к развитию эмфиземы легких, частых инфекционных заболеваний органов дыхания и дыхательной недостаточности.

Буллезная эмфизема может быть причиной спонтанного пневмоторакса у пациентов с синдромом Марфана
Кожа и мягкие ткани
Встречается повышенная растяжимость кожного покрова, которая сочетается с развитием атрофических стрий. Последние появляются спонтанно, они никак не связаны с колебанием веса, беременностью или гормональными нарушениями. Подкожный жир выражен слабо у пациентов с синдромом Марфана. Они часто страдают рецидивирующими грыжами передней брюшной стенки.
Встречаются и многие другие патологические симптомы поражения прочих органов и тканей при СМ. Например, опущение почек (нефроптоз), выпадение мочевого пузыря и матки у женщин, варикозное расширение вен, хронические запоры и др.

Диагностика синдрома Марфана
Диагностика при синдроме Марфана носит в основном клинический характер. Обязательно учитывают анамнез, в том числе и семейный (наличие подобных проблем у кого-то из родственников), данные объективного обследования и осмотра. Также проводят множество дополнительных диагностических процедур для выявления патологии тех или иных органов и систем. Для этого применяют ЭКГ, УЗИ сердца и сосудов, рентгенографию органов грудной клетки, КТ, МРТ внутренних органов, позвоночника, головного мозга, офтальмоскопию и прочие исследования органа зрения, аортографию, ангиографию и много других методик, в зависимости от клинической ситуации и симптомов болезни.
Существуют общепринятые диагностические критерии синдрома Марфана (большие и малые), которые позволяют с большой степенью вероятности поставить правильный, но предварительный диагноз пациенту.

Окончательный диагноз синдрома Марфана выставляют только после анализа генотипа (ДНК-диагностика) и выявления специфической мутации в гене, ответственном за продукцию фибриллина, с помощью молекулярно-генетических методик.
Лечение заболевания
К сожалению, на сегодняшний день вылечить синдром Марфана, как и повлиять на его причину, невозможно. Терапия в основном направлена на улучшение качества жизни больного человека, устранение симптомов и профилактику осложнений.
Лечение синдрома Марфана должно быть комплексным и может включать как консервативные, так и хирургические методики.
Всем пациентам с СМ рекомендуют ограничения в физической активности к среднему или низкому уровню, избегать тяжелого физического труда, не заниматься спортом, так как это способствует прогрессированию патологии и травматизму.
Больные должны наблюдаться у различных докторов: кардиолога, окулиста, травматолога-ортопеда, клинического генетика, невролога и др.

Лечением синдрома Марфана должна заниматься целая команда специалистов
В случае выявления тех или иных кардиоваскулярных проблем назначают комплексное лечение, медикаменты для профилактики осложнений. Проводят медикаментозную коррекцию аритмий, частоты сердечных сокращений, артериального давления. Обязательно всем пациентам назначают прием бета-блокаторов, если нет противопоказаний к этой группе средств, которые имеют протекторный эффект в отношении возможного расслоения аорты.
В случае патологии клапанного аппарата сердца, аневризме аорты и других сосудов, расслоении их стенок может понадобиться операция. Также хирургическое вмешательство используют для коррекции деформаций опорно-двигательного аппарата.
Рекомендуемые специалистами схемы терапии больных с СМ в обязательном порядке включают коллаген-образующие и метаболические средства, препараты для восполнения макро- и микроэлементов.
В случае проблем с органом зрения также проводят оперативные вмешательства при эктопии хрусталика, лазерную коррекцию зрения, подбирают очки или контактные линзы.
В целом подбор лечебных методик и спектр применяемых средств очень индивидуальны. Это полностью зависит от присутствующих симптомов и степени выраженности болезни у конкретного пациента.

Основная задача в лечении пациентов с Синдромом Марфана заключается в предотвращении сердечно-сосудистых повреждений
Прогноз
Синдром Марфана отличается, как правило, хроническим прогрессирующим течением. Продолжительность жизни пациентов при условии полноценного комплекса лечебных мероприятий в среднем составляет 45 лет. Основными факторами риска преждевременной смерти выступают осложнения, которые возникают вследствие патологии кардиоваскулярной системы.
Синдром Марфана и беременность
Пациентки с СМ могут иметь детей, причем здоровых, но это очень опасно по двум причинам:
- Беременная женщина имеет очень высокий риск летальных осложнений СМ со стороны сердечно-сосудистой системы. Так как при вынашивании ребенка создается усиленная нагрузка на организм матери, а особенно на сердце и сосуды, то очень большие шансы у пациенток с синдромом Марфана получить разрыв аневризмы, ее формирование, расслоение аорты и прочие смертельно опасные осложнения.
- Риск передачи данной наследственной патологии своему ребенку составляет 50%.
Поэтому специалисты не рекомендуют беременеть женщинам с синдромом Марфана.
Возможна ли профилактика болезни?
К сожалению, специфической профилактики синдрома Марфана не существует. Семейная пара, в которой один или оба родителя страдают данной патологией, в обязательном порядке должны планировать беременность и пройти медико-генетическое консультирование у врача-генетика. Вместе с этим можно проводить пренатальную (дородовую) диагностику на предмет наличия СМ у плода.
Источник