
Врожденный нефротический синдром финского типа

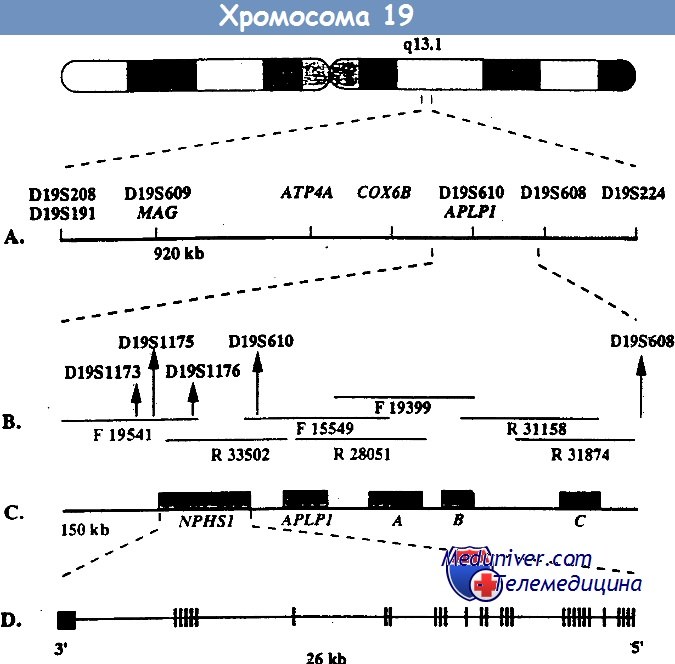
Врожденный нефротический синдром финского типа – клиника, диагностикаВрожденный нефротический синдром финского типа – заболевание наследуется по аутосомно-рецессивному типу и является основной причиной высокой протеинурии у детей первого месяца жизни. Хотя наибольшая распространенность этого заболевания отмечается в Финляндии (1,2 случая на 10 000 беременностей), описано много случаев заболевания у детей других национальностей. При этом заболевании протеинурия возникает еще внутриутробно, что проявляется повышенным уровнем а-фетопротеина в околоплодных водах. Уже на первой неделе жизни часто возникают отеки. Истощение, тяжелые инфекции и тромбозы обусловливают тяжесть заболевания и высокую смертность, ранее больные погибали на первом году жизни. Сегодня при интенсивном лечении больные могут дожить до того момента, когда им можно провести трансплантацию почки. Выживаемость как трансплантата, так и больных очень высокая. Локус, мутация в котором обусловливает данное заболевание, был найден с помощью позиционного клонирования на длинном плече 19-й хромосомы (19q13.1) и в финских, и в других семьях. При определении нуклеотидной последовательности этого локуса был найден ранее неизвестный ген NPHS1, который избирательно экспрессируется в подоцитах. Продукт этого гена получил название нефрин. Он относится к молекулам адгезии из суперсемейства иммуноглобулинов. Нефрин локализован в области щелевых диафрагм — видоизмененных плотных контактов между отростками ножек подоцитов. У больных с мутацией гена NPHS1 нет отростков ножек подоцитов и щелевых диафрагм. Это позволяет думать, что именно нефрин является важнейшим компонентом щелевых диафрагм, предотвращающих выход белка из сосудов клубочка. Среди всех мутаций гена NPHS1 у финнов преобладают две: Fin-major и Fin-minor. Они присутствуют более чем у 90% больных. Мутация Fin-major вызвана делецией двух пар нуклеотидов во 2-м экзоне, который кодирует терминирующий кодон, она встречается примерно у 80% больных финнов. Мутация Fin-minor — нонсенс-мутация в 26-м экзоне, она встречается примерно у 17% больных финнов. У больных других национальностей встречаются различные мутации по типу делеций, вставок, нонсенс- и миссенс-мутаций, а также мутации, нарушающие сплайсинг. Врожденный нефротический синдром финского типа — основная, но не единственная причина нефротического синдрома на первом месяце жизни.
– Также рекомендуем “Синдром Дени-Дрэша – клиника, диагностика” Оглавление темы “Наследственные болезни почек”:
|
Источник
Вторичный и врожденный нефротические синдромы у детей. Диагностика
Нефротический синдром может быть также вторичным проявлением многих поражений почечных клубочков: мембранозная нефропатия, мезангиокапиллярный, постинфекционный и волчаночный гломерулонефрит, геморрагический васкулит. Вторичный нефротический синдром следует подозревать у больных старше 8 лет при наличии артериальной гипертонии, гематурии, нарушения функции почек, внепочечных симптомов (сыпь, артралгия и др.) или низкого уровня комплемента в крови.
В некоторых регионах мира ведущими причинами нефротического синдрома являются малярия и шистосомоз. Вызывают нефротический синдром вирусы гепатита В и С, филярии, возбудитель проказы и ВИЧ.
Нефротический синдром сопутствует злокачественным опухолям, особенно у взрослых. У больных с солидными опухолями (например раком легкого, желудка или кишечника) почечная патология часто напоминает мембранозную нефропатию. По видимому, в этих случаях в почках откладываются комплексы опухолевых антигенов со специфическими антителами. При лимфомах, особенно лимфоме Ходжкина, поражение почек чаще всего напоминает болезнь минимальных изменений. Предполагается, что продуцируемый лимфомой лимфокин увеличивает проницаемость стенки клубочковых капилляров. Нефротический синдром возможен и до обнаружения опухоли, но исчезает при ее регрecce и возобновляется при рецидиве.
К развитию нефротического синдрома приводит также многочисленные лекарственные средства и химические вещества. Гистологическая картина почек в этих случаях может напоминать мембранозную нефропатию (пенициллин, каптоприл, препараты золота, НПВС, соединения ртути), болезнь минимальных изменений (пробенецид, этосуксимид, метимазол, литий) или мезангиокапиллярный гломерулонефрит (прокаинамид, хлорпропамид, фенитоин, триметадион, параметадион).
Врожденный нефротический синдром у детей
При развитии нефротического синдрома у детей первых 3 мес. жизни он считается врожденным. Чаще всего наблюдается врожденный нефротический синдром финского типа — аутосомно-рецессивное заболевание, наиболее распространенное среди потомков выходцев из Скандинавии (частота 1:8000). В его основе лежит мутация расположенного на хромосоме 19 гена NPHS1, который кодирует белок нефрин (важнейший компонент фильтрационных щелей между ножками подоцитов). Считается, что нефрин определяет нормальную функцию фильтрационного барьера в почечных клубочках. Гистологически синдром характеризуется в основном расширением проксимальных канальцев, пролиферацией клеток мезангия и склерозом клубочков.
У грудных детей обнаруживаются массивная протеинурия (которую можно определить и внутриутробно по повышению уровня а-фетопротеина) и отеки. Плацента увеличена. Больные часто рождаются недоношенными с респираторным дистрессом и расхождением швов черепа. Отеки сохраняются, часто возникают инфекционные заболевания, и к 5-летнему возрасту больные обычно погибают от почечной недостаточности. Кортикостероиды и иммуносупрессивные средства неэффективны.
Уменьшить протеинурию и улучшить состояние больных можно с помощью ингибиторов АПФ, индометацина и односторонней нефрэктомии. Однако в этих случаях чаще выполняют двустороннюю нефрэктомию с последующим постоянным диализом и активными диетическими мерами. В конце концов требуется трансплантация почки. В семьях, где имелись или имеются больные с врожденным нефротическим синдромом финского типа, для пренатальной диагностики определяют уровень а-фетопротеина в амниотической жидкости. Диагноз можно подтвердить данными анализа ДНК.
Другие причины врожденного нефротического синдрома включают врожденные инфекции, такие как сифилис, токсоплазмоз, краснуха и ЦМВ-инфекция, а также ВИЧ и вирус гепатита В. Поражение почек в этих случаях выражено слабее, чем при врожденном нефротическом синдроме финского типа, и излечение основного заболевания смягчает или устраняет почечную патологию.
У небольшого числа больных с врожденным нефротическим синдромом наблюдается диффузный мезангиальный склероз с прогрессирующим склерозом мезангия клубочков и быстрым ухудшением функции почек. Терминальная стадия почечной недостаточности развивается за несколько месяцев или лет. Диффузный мезангиальный склероз может быть как отдельным заболеванием, так и проявлением синдрома Дени-Дрэша (сочетание нефробластомы с мужским псевдогермафродитизмом). В основе этого синдрома лежит мутация гена опухоли Вильмса (WT1) на хромосоме 11.
– Также рекомендуем “Функция канальцев почек. Канальцевая фильтрация”
Оглавление темы “Заболевания почек у детей”:
- Нефротический синдром у детей. Причины и диагностика
- Идиопатический нефротический синдром у детей. Диагностика
- Лечение нефротического синдрома у детей. Гормоны
- Осложнения нефротического синдрома у детей. Прогноз
- Вторичный и врожденный нефротические синдромы у детей. Диагностика
- Функция канальцев почек. Канальцевая фильтрация
- Почечный канальцевый ацидоз. Проксимальноканальцевый
- Цистиноз у детей. Синдромы Лоу и Фанкони
- Дистальноканальцевый ацидоз у детей. Клиника
- Гиперкалиемический некроз почечных канальцев у детей. Диагностика и лечение
Источник
Врожденный нефротический синдром финского типа(Finnish Type N.S., finnisher Type NS, неонатальный нефроз) был описан R.Norio в 1966 г. Заболевание часто встречается в Финляндии — 1:8200 рождений [banning P. et al., 1989]. Врожденный нефроз регистрируется в Северо-Западном регионе России, причем не всегда устанавливается этническая зависимость. Это аутосомно-рецессивное заболевание. Предполагается, что ген локализован на 19-й хромосоме.
Патогенез врожденного НС финского типа остается доконца не известен. При морфологическом исследовании обнаруживают микрокистоз проксимальных канальцев в корти-ко-медуллярной зоне, признаки незрелости клубочков. Обычная светооптическая и электронная микроскопия не выявляют изменений базальной мембраны.
Заболевание проявляется полным клинико-лабораторным симптомокомплексом НС, нередко с гематурией, в первые дни и недели (до 3 мес) после рождения. Выраженные отеки отмечаются у детей уже при рождении. Большинство авторов указывают на токсикоз беременных, преждевременные роды, большую отечную плаценту, окрашенные меконием околоплодные воды, малую массу тела новорожденных. Масса плаценты достигает 25—50% от массы тела новорожденного. Врожденный НС финского типа является гормонорезистентным, с неблагоприятным прогнозом.
C.Holmberg и соавт. (1995) рекомендуют детям с финским типом НС внутривенные инфузии альбумина 3—4 г/кг с последующим струйным введением лазикса 0,5 мг/кг. Авторы считают необходимым назначение витамина D2 (2000 МЕ/сут), а также магнезии 40—60 мг/сут, кальция, проведение профилактики инфекционных и тромботических осложнений.
При активной консервативной терапии дети с финским типом врожденного НС достигают возраста, в котором возможны постоянный перитонеальный диализ и трансплантация почки.
Прогноз ВНС финского типа остается серьезным. Летальный исход возможен уже на первом году жизни в результате вторичной вирусно-бактериальной инфекции, гиповолемических, тромботических осложнений, отека мозга, кахексии.
Врожденный нефротический синдром французскоготипа (French Type, franzosicher N.S.) Врожденный НС французского типа передается аутосомно-рецессивно. Симптомокомплекс НС диагностируют на 1 —12-й неделе жизни ребенка. При микроскопии устанавливают диффузный мезангиальный, а затем и глобальный склероз без существенной клеточной пролиферации. При электронной микроскопии выявляют исчезновение ножек подоцитов. Врожденный НС с мезангиальным склерозом характеризуется гормонорезистентностью и неблагоприятным прогнозом с исходом в почечную недостаточность к 1 — 1,5 годам. Трансплантация почки продлевает жизнь обреченных детей.
Врожденный нефротический синдром с минимальными изменениями.В большинстве случаев врожденного НС с минимальными изменениями наблюдают гормоночувствительные варианты. Многие авторы отмечают полную ремиссию при НСМИ после глюкокортикоидной терапии или спонтанную ремиссию.
Прогноз врожденного НС с минимальными клубочковыми изменениями может быть благоприятным и неблагоприятным вследствие развития серьезных осложнений.
Врожденный нефротический синдром с морфологической картиной мезангиопролиферативного гломерулонефрита. Наблюдалась спонтанная ремиссия врожденного НС с мезангиопролиферативными изменениями. Однако J.Wiggelinkhuizen и соавт. (1972) наблюдали у ребенка 3 мес врожденный НС с морфологической картиной мезангиопролиферативного ГН и, несмотря на лечение кортикостероидами и циклофосфамидом, с не столь благоприятным прогнозом.
Врожденный нефротический синдром с фокально-сегментарным гломерулосклерозом.Описан случай врожденного гормонорезистентного НС с ФСГС. Лечение преднизолоном и циклофосфамидом, гаммаглобулином оказалось малоэффективным, сохранялась персистирующая протеинурия.
Инфантильный нефротический синдром с минимальными изменениями (возникший у грудных детей).На первом году жизни у детей, чаще после 6—9 мес, может наблюдаться дебют полного симптомокомплекса НС, без артериальной гипертензии, гематурии, нарушения функции почек, с минимальными изменениями гломерул, так называемого НСМИ — липоидного нефроза.
При НСМИ важное патогенетическое значение отводится изменениям анионных участков (потеря отрицательного заряда на lamina гага externa гломерулярной базальной мембраны, подоцитах). Это приводит к утрате зарядно-селективной функции клубочкового фильтрационного барьера и возникновению протеинурии. По ответу на глюкокортикоидную терапию встречаются гор-моночувствительные, зависимые или резистентные варианты инфантильного НСМИ. Течение НСМИ, возникшего на первом году жизни ребенка, — рецидивирующее и часто рецидивирующее. Возможно наступление стойкой ремиссии после глюкокортикоидной терапии дебюта заболевания. Прогноз инфантильного НСМИ — благоприятный. Длительная ремиссия и отсутствие нарушения функции почек свидетельствуют о клиническом выздоровлении.
Инфантильный НС с мезангиопролиферативным ГН.У детей с 4 до 12 мес может возникнуть инфантильный НС с атопией (экзема, аллергический ринит, астма), морфологически классифицируемый как мезангиально-пролиферативный ГН.
В терапии инфантильного НС с мезангиально-пролифера-тивным ГН применяют преднизолон 7-10 мес, при гормоно-резистентности — преднизолон и циклоспорин А 12 мес с положительным эффектом.
При адекватной терапии прогноз инфантильного НС с атопией благоприятный.
Инфантильный нефротический синдром с ФСГС.Инфантильный НС с ФСГС проявляется у младенцев с 4 до 12 мес. Как правило, наблюдаются резистентность к глюкокортикоидной терапии и неблагоприятный прогноз.
Инфантильный нефротический синдром с мембранозным ГН.Идиопатическая мембранозная нефропатия у детей первого года жизни описана J.D.Mahan и соавт. (1988), T.Jo.Mauch и соавт. (1993) и приведена в ряде классификаций J.Rapola и соавт. (1991), T.Jo.Mauch и соавт. (1993).
Источник
Терминология. Под врожденным нефротическим синдромом понимается НС, развившийся у ребенка до 3-месячного возраста. Врожденный HC может быть первичным, генетически детерминированным и вторичным при врожденной цитомегалии, токсоплазмозе, сифилисе, туберкулезе, тромбозе почечных вен, СПИДе. Особое место среди врожденного HC занимает первичный наследственный, так называемый врожденный нефротический синдром финского типа. Это аутосомно-рецессивно наследуемая патология, проявляющаяся с первых дней жизни ребенка тяжелым нефротическим синдромом с большой протеинурией и резкой гипопротеи-немией. При «естественном» течении летальный исход наступает до 1 года, причем к нему приводят либо развитие почечной недостаточности, либо септические осложнения.
История и эпидемиология. Впервые заболевание описано в 1966 г. R. Norio. При анализе церковно-приходских книг в юго-западном регионе Финляндии, где наиболее часто встречалось это заболевание, был обнаружен родоначальник патологии — финн, который проживал в этом регионе во второй половине XVI в. До проведения антенатальной диагностики заболевание встречалось с частотой 1:8200 рождений. Аналогичные случаи регистрируются в северо-западном районе России, в Ленинградской области. He всегда удается подтвердить этническую (финскую) принадлежность семьи. Этот вариант патологии неоднократно описывался в различных странах мира у лиц нефинской национальности.
Клиническая характеристика. Течение беременности тяжелое, роды, как правило, преждевременные, масса плаценты составляет более 1/4-1/2 массы новорожденного. Чаще ребенок рождается уже с выраженными отеками, но они могут появиться несколько позднее — к концу первого месяца жизни. Протеинурия достигает 10 г за сутки. Резко выражена гипоальбуминемия, имеет место повышение липидов сыворотки крови. При уменьшении отечного синдрома после введения диуретиков обращают на себя внимание резкая дистрофия ребенка, множественные стигмы дизэмбриогенеза. Резко снижены показатели иммунной защиты, что является основой развития гнойных осложнений. Возможны тромбоэмболии. АД снижено или в пределах нормы. В амниотической жидкости и сыворотке крови беременных в высоком титре содержится альфа-фетопротеин. Обнаружение этого феномена позволило проводить своевременную антенатальную диагностику.
Морфология и патогенез. При гистологическом исследовании почек обнаруживаются микрокистоз проксимальных канальцев в кортикомедуллярной зоне, мультигломерулярность и другие признаки незрелости почечной ткани, пролиферация мезангиальных клеток, фиброзные изменения.
Врожденный нефротический синдром финского типа относится к гломерулярным болезням, причем продукт гена — нефрин — локализован на подоцитах. Недостаточность нефри-на вызывает протеинурию еще в антенатальном периоде развития ребенка.
Генетика. Врожденный HC финского типа наследуется аутосомно-рецессивным путем. М. Kestila и соавт. при исследовании 17 семей с указанной патологией не обнаружили дефекта ни в одном из генов альфа-1-, альфа-2-, альфа-3- и альфа-4-цепи коллагена IV типа, а также основных генов цепей ламинина и гепа-рансульфат-протеогликана, кодирующих основные компоненты БМ клубочков. Получены убедительные данные, что мутантный ген локализован на 19ql3, этот ген — NPHSI — кодирует трансмембранный протеин — нефрин, присущий подоцитам.
Современными исследованиями выяснено, что в различных регионах мира, где выявлялся врожденный НС, близкий по сути финскому, имеется около 40 мутаций гена NPHSI. Однако в Финляндии у больных и носителей обнаружены только 2 идентичные мутации этого гена. В семьях, где имеется врожденный НС, в процессе медико-генетического консультирования беременные женщины обязательно обследуются на наличие у них в крови альфа-фетопротеина. При его обнаружении рекомендуется прерывание беременности.
Диагностика. Рождение в семье ребенка с врожденным HC требует прежде всего выяснения этнических корней. Обязательно исключение вторичного НС, связанного с внутриутробными инфекциями. Врожденный HC финского типа следует дифференцировать от семейного НС, который описан в различных странах мира у людей различной национальности (см. ниже). О финском типе HC говорят тяжелая беременность, наличие очень крупной плаценты, обнаружение при морфобиоптическом исследовании микрокистоза проксимальных канальцев.
Лечение. Несмотря на то что в Финляндии проводится активное выявление семей, где возможно развитие врожденного HC финского типа, все же и в настоящее время рождаются дети с этой тяжелой патологией. Ни симптоматическая терапия, ни стероиды и иммуносупрессоры не вызывают улучшения у больных с врожденным HC финского типа.
Рекомендуется высокобелковая и высококалорийная диета наряду со строжайшим водноэлектролитным сбалансированным режимом до 10—12 мес жизни ребенка. К этому возрасту удается довести его массу тела до 10 кг, ликвидировать дистрофию и отечный синдром. После нефрэктомии проводят почечную трансплантацию. Десятилетнее наблюдение за группой, включавшей около 40 детей, убедительно свидетельствует о хорошей реабилитации таких больных.
Источник