
Болезнь краббе код по мкб

Текущая версия страницы пока не проверялась опытными участниками и может значительно отличаться от версии, проверенной 17 марта 2013;
проверки требуют 17 правок.
Боле́знь Кра́ббе (галактозилцерамидный липидоз[1][2]) — редкое наследственное заболевание из группы лизосомных болезней накопления[1][2], при котором поражается миелиновая оболочка нервных волокон. Оно обусловлено дефицитом фермента – галактозидазы цереброзидов, осуществляющей второй этам катаболизма сульфатидом; накапливаются галактозилцерамид и галактозилсфингозин.[3] Наследуется аутосомно-рецессивно. Болезнь названа в честь датского невролога Краббе (Knud Haraldsen Krabbe), который описал её в 1916 году[4].
Эпидемиология[править | править код]
Частота болезни Краббе составляет 1 на 100000 новорождённых в США[5], 2 на 100000 — в скандинавских странах[6] и самая высокая среди палестинских арабов — 1 на 6000[7]. Заболевание также встречается у кошек[8] и собак, особенно у вест-хайленд-уайт-терьеров и керн-терьеров[9][10].
Этиология[править | править код]
Болезнь Краббе вызвана мутациями в GALC гене, расположенного на 14 хромосоме (14q31)[11], которые приводят к недостаточности фермента галактоцереброзидазы. В результате в бимолекулярном слое миелина происходит накопление предшественника галактоцереброзида — галактозилцерамида (психозина), который вызывает гибель олигодендроцитов и распад миелинового волокна с образованием характерных включений — глобоидных клеток[12].
Клиническая картина[править | править код]
Болезнь Краббе обычно начинает проявляться в 3-6 месяцев жизни. Первыми симптомами бывают немотивированное повышение температуры тела, гипервозбудимость, нарушения вскармливания, ригидность конечностей, судороги, рвота, отставание в психомоторном развитии. В дальнейшем мышечный тонус нарастает до опистотонуса, происходит прогрессирующее снижение интеллекта и зрения. Поздние формы заболевания (ювенильная и взрослая) характеризуются более медленным прогрессированием.
Диагностика[править | править код]
При морфологическом исследовании определяется скопление шарообразных крупных многоядерных клеток (глобоидных) в участках демиелинизации.
Проводят магнитно-резонансную томографию с целью уточнения локализации и специфики поражения.[3]
Прогноз[править | править код]
Новорождённые с болезнью Краббе погибают обычно в течение 2-х лет. При поздних формах заболевания с медленным прогрессированием продолжительность жизни значительно больше.
Лечение[править | править код]
Трансплантация костного мозга на ранних стадиях заболевания позволяет снизить тяжесть заболевания и уменьшить клинические проявления. Публикация в Медицинском журнале Новой Англии от 2005 года сообщает, что трансплантация пуповинной крови успешно останавливает болезнь, но только в том случае, если проводится до появления явных симптомов[13].
Примечания[править | править код]
- ↑ 1 2 Т. Р. Харрисон. Внутренние болезни в 10 книгах. Книга 8. Пер. с англ. М., Медицина, 1996, 320 с.: ил. Глава 316. Лизосомные болезни накопления (с. 250—273). med-books.info. Дата обращения 14 ноября 2014.
- ↑ 1 2 Справочник Т. Р. Харрисона по внутренним болезням, 1992—1997:. Глава 316. Лизосомные болезни накопления. rusmedserver.ru. Дата обращения 14 ноября 2014.
- ↑ 1 2 Л.О.Бадалян. Детская неврология. — “Медицина”, 2010.
- ↑ Whonamedit — Krabbe’s disease
- ↑ Krabbe disease. Genetics Home Reference. United States National Library of Medicine (2 мая 2008). Дата обращения 7 мая 2008. Архивировано 16 августа 2012 года.
- ↑ Books.Google.com
- ↑ Zlotogora J. Autosomal recessive diseases among palestinian Arabs (англ.) // Journal of Medical Genetics (англ.)русск. : journal. — 1997. — September (vol. 34, no. 9). — P. 765—766. — ISSN 0022-2593. — doi:10.1136/jmg.34.9.765. — PMID 9321766.
- ↑ Salvadori C., Modenato M., Corlazzoli D.S., Arispici M., Cantile C. Clinicopathological features of globoid cell leucodystrophy in cats (англ.) // J. Comp. Pathol. : journal. — 2005. — May (vol. 132, no. 4). — P. 350—356. — doi:10.1016/j.jcpa.2004.12.001. — PMID 15893994.
- ↑ NYtimes.com
- ↑ Capucchio M.T., Prunotto M., Lotti D., Valazza A., Galloni M., Dore B., Pregel P., Amedeo S., Catalano D., Cornaglia E., Schiffer D. Krabbe’s disease in two West Highland White terriers (англ.) // Clin. Neuropathol. : journal. — 2008. — Vol. 27, no. 5. — P. 295—301. — PMID 18808060.
- ↑ Regional mapping of the human galactocerebrosidase gene (GALC) to 14q31 by in situ hybridization. doi:10.1159/000133703. Архивировано 16 августа 2012 года.
- ↑ Неврология. Национальное руководство. — ГЭОТАР-Медиа, 2010. — С. 872-873. — 2116 с. — 2000 экз. — ISBN 978-5-9704-0665-6.
- ↑ Escolar M.L., Poe M.D., Provenzale J.M., Richards K.C., Allison J., Wood S., Wenger D.A., Pietryga D., Wall D., Champagne M., Morse R., Krivit W., Kurtzberg J. Transplantation of Umbilical-Cord Blood in Babies with Infantile Krabbe’s Disease (англ.) // New England Journal of Medicine : journal. — 2005. — Vol. 352, no. 20. — P. 2069—2081. — ISSN 0028-4793. — doi:10.1056/NEJMoa042604. — PMID 15901860.
Источник
Что такое болезнь Краббе?
Болезнь Краббе (или сфинголипидоз, тип Краббе, галактозилцерамидный липидоз) является редким наследственным нарушением обмена липидов, вызванным дефицитом фермента галактоцереброзидазы (GALC), который необходим для расщепления (метаболизма) сфинголипидов галактозилцеремида и психозина.
Неспособность разрушить эти сфинголипиды приводит к дегенерации миелиновой оболочки, окружающей нервы головного мозга (т.е. к демиелинизацие). В пораженных участках головного мозга появляются характерные глобоидные клетки. Это метаболическое нарушение характеризуется прогрессирующей неврологической дисфункцией, такой как умственная отсталость, паралич, слепота, глухота и паралич определенных лицевых мышц (псевдобульбарный паралич). Галактозилцерамидный липидоз наследуется как аутосомно-рецессивное расстройство.
Признаки и симптомы

Начало болезни Краббе в преобладающей инфантильной форме (90% случаев) происходит в возрасте от одного до семи месяцев. Поздняя форма расстройства возникает в возрасте 18 месяцев или старше, включая подростковый возраст и зрелость.
Специфические симптомы и степень болезни Краббе варьируются от случая к случаю. Младенцы, пораженные заболеванием, могут быть беспокойными и чрезмерно раздражительными (повышенная возбудимость). Рвота, необъяснимые лихорадки и частичная потеря сознания являются дополнительными возможными симптомами. Нижние конечности могут иметь спастические сокращения. Также могут возникать судороги, характеризующиеся чередованием сокращения и расслабления (клоническое) или постоянным напряжением (тоническое). Пострадавшие дети гиперчувствительны к различным раздражителям, таким как звуки.
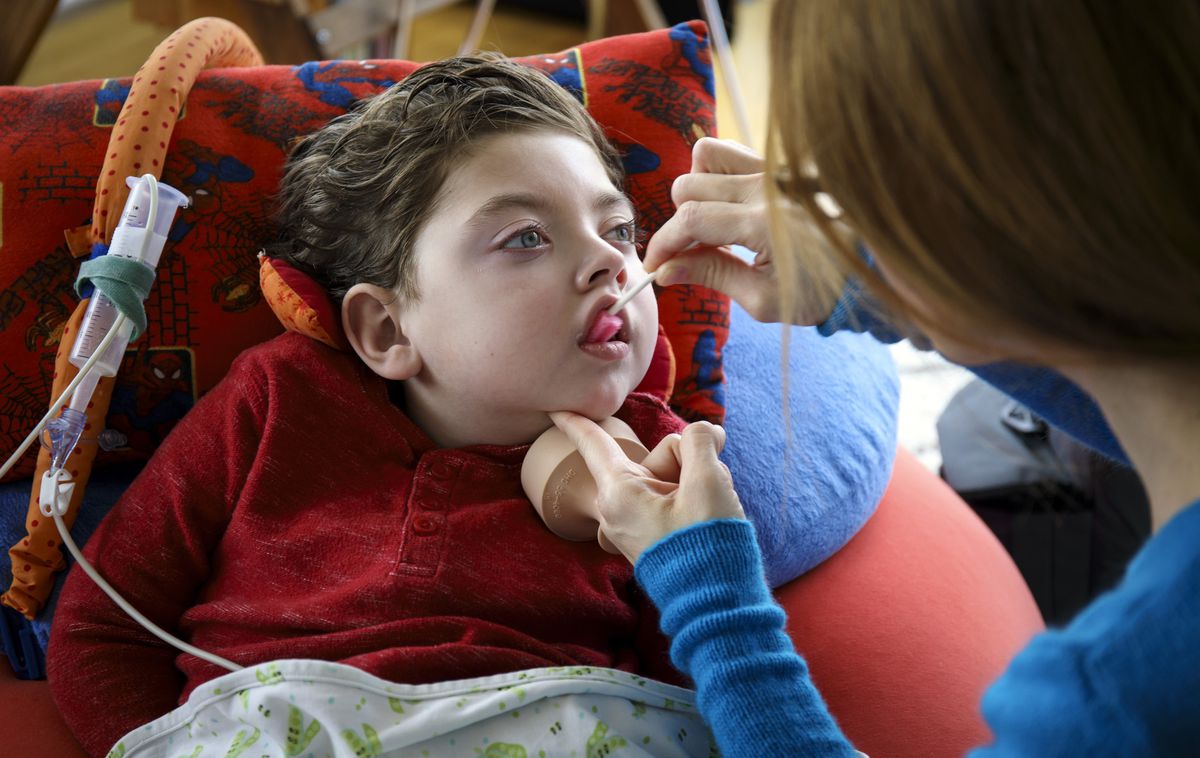
Умственное и физическое развитие может быть медленным. В некоторых случаях может произойти регресс ранее приобретенных навыков. Из-за дегенерации определенных частей мозга ноги иногда жестко вытянуты в бедре и колене; руки могут вращаться в плече и вытягиваться в локте; лодыжки, пальцы ног и пальцы могут быть согнуты. Также может возникнуть слепота, вызванная дегенерацией коры головного мозга. У людей с галактозилцерамидным липидозом также могут быть проблемы с глотанием (дисфагия) и периферическая нейропатия, состояние, характеризующееся мышечной слабостью; болью; онемением; покраснением; и/или жжением или покалыванием в пораженных областях, особенно в руках и ногах (конечностях). ЛРасстройства часто приводит к опасным для жизни осложнениям.
При ювенильной и взрослой формах болезни Краббе первоначальным симптомом может быть нарушение контроля произвольных движений и прогрессирующая ригидность мышц ног (спастический парапарез). Пострадавшие люди с этими формами расстройства могут также испытывать прогрессирующую потерю зрения и заболевание, затрагивающее множественные нервы (полинейропатия).
Причины
Галактозилцерамидный липидоз — это наследственное заболевание, передающееся потомству через рецессивные гены. Оно вызвано дефицитом фермента галактозид-бета-галактозидазы (галактозилкерамидазы). Этот фермент необходим для метаболизма галактоцереброзида (галактозилкерамида), компонента жировой оболочки вокруг нервов (миелина). Демиелинизация нервных клеток в больших полушариях мозга (и в стволе мозга) вызывает неврологические симптомы болезни Краббе.
Человеческие черты, включая классические генетические заболевания, являются продуктом взаимодействия двух генов для данного состояния, один из которых получается от отца, а другой — от матери. При рецессивных расстройствах состояние не появляется, если человек не наследует один и тот же дефектный ген по одному признаку от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, человек будет носителем заболевания, но обычно бессимптомным. Риск передачи заболевания детям пары, которые являются носителями рецессивного расстройства, составляет 25 процентов. 50 процентов их дети рискуют быть носителями этого заболевания, но обычно не проявлять симптомов расстройства. 25 процентов их дети могут получить оба нормальных гена, по одному от каждого родителя, и быть генетически нормальным (для этой конкретной черты). Риск одинаков для каждой беременности.
Исследователи установили, что болезнь Краббе может быть вызвана нарушением или изменениями (мутациями) гена галактоцереброзидазы человека (GALC), расположенного на длинном плече (q) хромосомы 14 (14q31). Хромосомы находятся в ядре всех клеток организма. Они несут генетические характеристики каждого человека. Пары человеческих хромосом пронумерованы от 1 до 22, с неравной 23-й парой Х и Y хромосом для мужчин и двумя Х хромосомами для женщин. Каждая хромосома имеет короткую руку, обозначенную буквой «р», и длинную руку, обозначенную буквой «q». Хромосомы далее подразделяются на полосы, которые пронумерованы. Например, «хромосома 14q31» относится к полосе 31 на длинном плече хромосомы 14.
Затронутые группы населения
Примерно 1 из 100 000 новорожденных страдает болезнью Краббе. Мужчины страдают так же часто, как женщины.
Близкие по симптомам расстройства
Симптомы следующих расстройств могут быть похожи на симптомы болезни Краббе. Сравнения могут быть полезны для дифференциальной диагностики:
- Адренолейкодистрофия (АЛД, или болезнь Шилдера) является одной из многих различных лейкодистрофий. Расстройство может проявляться в двух различных генетических формах: половой и неонатальной АЛД. Для обоих характерно разрушение липидных оболочек, окружающих нервы (демиелинизация) в головном мозге. Однако они различаются по типу наследования, степени тяжести и типу симптомов. Все типы АЛД характеризуются накоплением жирных кислот с очень длинной цепью (VLCFA), которые являются типом молекулы жира, накапливающего в тканях организма, особенно в надпочечниках и белом веществе мозга. Может также происходить накопление лимфатических и плазматических клеток вокруг кровеносных сосудов в центральной нервной системе.
- Болезнь Канавана (губчатая дегенерация головного мозга) является формой лейкодистрофии, которая вызывает замещение белого вещества головного мозга микроскопическими заполненными жидкостью пространствами. Это расстройство наследственное и характеризуется структурными нарушениями и ухудшением двигательных, сенсорных и интеллектуальных функций. Похоже, чаще всего влияет на лиц восточноевропейского еврейского происхождения. Расстройство является прогрессирующим и дегенеративным. Симптомы могут включать прогрессирующее снижение умственных функций, сопровождающееся потерей мышечного тонуса, плохим контролем над головой, ненормально увеличенной головой (мегалоцефалией) и/или слепотой.
- Метахроматическая лейкодистрофия (МЛД или сульфатид липидоз), наиболее распространенная форма лейкодистрофии, является редким наследственным нейрометаболическим нарушением, затрагивающим белое вещество мозга (лейкоэнцефалопатия). Он характеризуется накоплением жирного вещества, известного как сульфатид (сфинголипид), в мозге и других частях тела (например, в печени, желчном пузыре, почках и/или селезенке). Жировое защитное покрытие на нервных волокнах (миелин) теряется из областей центральной нервной системы (ЦНС) из-за накопления сульфатида. Симптомы метахроматической лейкодистрофии могут включать судороги, эпилепсию, изменения личности, спастичность, прогрессирующую деменцию, двигательные нарушения, прогрессирующие до паралича, и/или нарушения зрения, приводящие к слепоте. Метахроматическая лейкодистрофия наследуется как аутосомно-рецессивный признак.
- Болезнь Александера является чрезвычайно редким, прогрессирующим неврологическим расстройством, которое обычно проявляется в младенчестве или раннем детстве. Однако реже описывались случаи, когда появление симптомов происходило в более позднем детстве или в подростковом возрасте (ювенильное начало) или, реже, в течение третьего-пятого десятилетий жизни (взрослое начало). У младенцев и детей младшего возраста, пораженных болезнью Александера, сопутствующие симптомы и признаки включают в себя неспособность расти и набирать вес с ожидаемой скоростью (неспособность развиваться); задержки в развитии определенных физических, умственных и поведенческих навыков, которые обычно приобретаются на определенных стадиях (психомоторная отсталость); и прогрессирующее увеличение головы (макроцефалия). Дополнительные симптомы обычно включают в себя внезапные эпизоды неконтролируемой электрической активности в мозге (эпилепсию), спастичность и прогрессирующее неврологическое ухудшение. В некоторых случаях наблюдается гидроцефалия. В большинстве случаев болезнь Александера возникает случайно по неизвестным причинам (спорадически), без семейного анамнеза заболевания. Считается, что в очень небольшом числе случаев расстройство затрагивает более одного члена семьи.
Диагностика
Болезнь Краббе можно диагностировать, проверив активность фермента галактоцереброзидазы (галактозилцерамидазы) в клетках фибробластов, полученных от младенца или плода с помощью амниоцентеза.
Стандартные методы лечения
Специального лечения болезни Краббе не существует. Лечение симптоматическое и поддерживающее. Генетическое консультирование может быть полезным для семей детей, затронутых этой болезнью.
Для младенцев, у которых уже развились симптомы болезни Краббе, в настоящее время не существует лечения, которое могло бы изменить течение заболевания. Поэтому лечение направлено на устранение симптомов и оказание поддерживающей помощи. Вмешательства могут включать следующее:
- Противосудорожные препараты для лечения эпилепсии.
- Препараты для облегчения мышечной спастичности и раздражительности.
- Физиотерапия для минимизации ухудшения мышечного тонуса.
- Пищевая поддержка, такая как использование трубки для доставки жидкости и питательных веществ непосредственно в желудок (питание через гастростому).
Вмешательства для детей старшего возраста или взрослых с менее тяжелыми формами заболевания могут включать:
- Физиотерапия для минимизации ухудшения мышечного тонуса.
- Трудотерапия для достижения как можно большей независимости в повседневной деятельности.
Трансплантация стволовых клеток
Гемопоэтические стволовые клетки — это специализированные клетки, которые могут развиваться во все типы клеток крови в организме. Эти стволовые клетки также являются источником микроглии, специализированных клеток, поедающих мусор, который живет в нервной системе. При болезни Краббе микроглия превращается в токсичные глобоидные клетки.
При трансплантации стволовых клеток донорские стволовые клетки доставляются в кровоток реципиента через трубку, называемую центральным венозным катетером. Донорские стволовые клетки помогают организму вырабатывать здоровую микроглию, которая может заполнять нервную систему и доставлять функционирующие ферменты GALC. Это лечение может помочь восстановить некоторую степень нормального производства и поддержания миелина.
Эта терапия может улучшить результаты у детей, если лечение начинается до появления симптомов, т.е., когда диагноз является результатом скрининга новорожденного. Современные данные свидетельствуют о том, что трансплантация стволовых клеток наиболее эффективна, когда начинается до достижения ребенком возраста 2 недель.
Исследования показали, что у детей с симптомами после трансплантации стволовых клеток может наблюдаться более медленное прогрессирование заболевания и улучшение качества и продолжительности жизни по сравнению с детьми, которые не получают трансплантацию стволовых клеток до появления симптомов. Тем не менее, у детей, у которых была трансплантация стволовых клеток до появления симптомов, все еще возникают значительные трудности с речью, ходьбой и другими моторными навыками в детстве.
Дети старшего возраста и взрослые с легкими симптомами болезни Краббе также могут извлечь пользу из этого лечения. Как и у младенцев, тяжесть симптомов во время трансплантации стволовых клеток влияет на результаты лечения.
Прогноз и осложнения
Нейродегенерация и ранняя смерть (<2-3 года) встречаются в большинстве детских случаев. У поздних младенцев/ювенильных пациентов заболевание обычно заканчивается летальным исходом через 2-7 лет после появления симптомов. Взрослые пациенты могут выживать через много лет после появления симптомов.
У детей с прогрессирующей болезнью Краббе может развиться ряд осложнений, включая инфекции и респираторные заболевания. На более поздних стадиях заболевания дети становятся нетрудоспособными, прикованы к постели и в конечном итоге переходят в вегетативное состояние.
Большинство детей, у которых развивается болезнь Краббе в младенчестве, умирают до 2 лет, чаще всего от дыхательной недостаточности или осложнений, связанных с неподвижностью и заметно сниженным мышечным тонусом. Дети, у которых заболевание развивается позднее в детстве, могут иметь более продолжительную продолжительность жизни, обычно от 2 до 7 лет после постановки диагноза.
Источник
Лейкодистрофия – группа заболеваний с поражением мозжечка, белого вещества, полушарий головного мозга с сохранностью корковых структур.
Нейродегенерация мозговой ткани сопровождается накоплением внутри спинного и головного мозга метаболические соединений, разрушающих миелин. Повреждение оболочки нейронов приводит к необратимым заболеваниям, сопровождающимся двигательными расстройствами, нарушением психомоторной функции, поражение слуха и зрении, эпилепсией, судорогами, неврологическими расстройствами, эпилептическими приступами.
Лейкострофии МРТ
Классификация по МКБ 10
Международная классификация болезней 10 пересмотра относит лейкодистрофии к сфинголипидозам – заболеваниям, сопровождающимся избыточным отложением патологических жиров (липидов). Код нозологии – «E 75».
Нарушения обмена ганглиозидов кодируются «GM 2»:
- Ювенильная форма;
- Лейкодистрофия взрослых;
- Болезнь Сендхоффа;
- Синдром Тея-Сакса.
Другие ганглиозидозы («E 75.1»):
- Муколипидоз IV;
- Ганлиозидозы GM3, GM1.
Другие сфинголипидозы («E 75.2»):
- Недостаточность сульфатазы;
- Метахроматическая лейкодистрофия;
- Болезнь Нимана-Пика;
- Синдром Краббе;
- Синдром Фабера;
- Болезни Фабри-Андерсона.
Неуточненный сфинголипидоз – «E 75.3». К категории относятся все формы этиологические факторы, которых установить не удалось. Липофусциноз нейронов – «E 75.4». Избыточное образование атипичных жировых части приводит к нарушению передачи нервных сигналов. Неклассифицированные состояния («E 75.5»):
- Болезнь Волмана;
- Холестероз Ван-Богарта-Шерера.
Дисбаланс метаболических соединений внутри головного мозга обеспечивает атипичную клинику.
Неуточненная болезнь накопления липидов – «E 75.6».
Международная классификация МКБ 10 принята во всем мире для унификации перечная нозологических форм. Стандартизации тактики лечения.
Виды лейкодистрофии
Перечень биохимических изменений, приводящих к лейкодистрофии мозжечка, стволовых структур головного и спинного мозга, не выявлен. Ученые считают патологию вариантом повреждения лизосом. Научные исследования не выявили ферменты, отвечающие за клинические проявления нозологии.
Лизосомальные виды лейкодистрофий:
- Галлерводена-Шпатца;
- Краббе;
- Пелициуса-Мерцбахера.
Большинство форм лейкодистрофий возникает в раннем возрасте, но обнаруживается патология и у взрослых. При всех разновидностях возникают неврологические и пирамидальные расстройства, экстрапирамидальная ригидность, демиелинизация нервных волокон. Перечень лабораторных изменений при лейкодистрофиях – увеличение белка, усиленный плеоцитоз.
Метахроматическая лейкодистрофия
Проявляется у взрослых после 21 года. Преимущественно встречается нозология у мужчин. Наследуется по аутосомно-рецессивному механизму. Метахроматическая лейкодистрофия головного мозга развивается постепенно. До выраженных клинических симптомов может пройти более двадцати лет. Особенности проявлений психоза:
- Забывчивость;
- Снижение академических возможностей;
- Неразумные действия;
- Странности поведения;
- Излишняя подозрительность.
Аналогичные клинические симптомы возникают при шизофрении. Присоединение неврологических симптомов мозжечковой атаксии, пирамидальных расстройств, неловкость движений пациента провоцирует психическую деградацию личности. Беспомощность, отсутствие контакта с окружающими людьми, прикованность к постели обеспечивает быстрое прогрессирование клиники из-за ряда метаболических изменений:
- Падение активности лейкоцитарных ферментов (арилсульфатазы A);
- Усиленное выделение сульфатидов с уриной;
- Дисбаланс проведения нервного импульса по поврежденным волокнам;
- Перераспределение пигментного вещества.
Метахроматическая лейкодистрофия у детей (Гринфилда) сопровождается судорогами, атаксией, нистагмом. Признаки терминальной стадии лейкодистрофии у детей:
- Децеребрационная ригидность;
- Бульбарные расстройства;
- Тетраплегия.
Причиной метахромного вида является избыточное скопление липидов. Патогенетическим механизмом формирования патологии является недостаточность фермента цереброзидсульфатазы. Развивается нозология позже форм Краббе или Тея-Сакса. Примерно в 5 лет у ребенка нарушается походка из-за повышенного тонуса мускулатуры. Постепенно утрачивается рефлекторная активность, иннервация сухожилий.
Клинические симптомы лейкодистрофии
Большинство видов возникает в детском возрасте. Сразу после рождения патологических изменений у ребенка не прослеживается. Через несколько месяцев или лет прослеживается неврологическая или психическая симптоматика, которая постепенно усугубляется.
Признаки ранних стадий лейкодистрофии:
- Патология зрения;
- Олигофрения;
- Мышечный спазм;
- Подергивания конечностей;
- Гипертонус;
- Тонические судороги;
- Признаки экстрапирамидальной патологии (шаткая походка);
- Падение интеллекта.
Множественные чувствительные расстройства, патология глотания, глухота диагностируются у дошкольников.
Симптомы лейкодистрофии мозга у грудничков второго года жизни:
- Замедленное психомоторное развитие (олигофрения);
- Патология походки.
Клинические проявления, начинающиеся с третьего года жизни:
- Потеря слуха и зрения;
- Гипертермический синдром;
- Тетраплегия;
- Гипертермия (повышение температуры).
Тяжелая симптоматика появляется через 10 лет после начала первичных изменений головного мозга.
Первичные изменения мозга сопровождаются спастичностью, миоклонией, задержкой развития, мышечным тремором. У взрослых прогрессирующая форма сопровождается быстрой потерей свойств личности, расстройствами речи, патологическим мышлением. Постепенное прогрессирование сопровождается разнообразными изменениями слизистой оболочки с развитием спастичности, мышечными судорогами, гипертонусами.
Вариант метахроматической лейкодистрофии сопровождается психозом, деменцией, эмоциональной неустойчивостью, расстройством речи, мышлением.
Томограммы метахроматической лейкодистрофии
Первые признаки лейкодистрофии у ребенка
При большинстве лейкодистрофий первые симптомы появляются на четвертом году жизни. Диагностировать нозологию удается по следующим признакам:
- Повышенный мышечный тонус;
- Сильная нервная возбудимость;
- Психомоторное развитие не соответствует возрасту;
- Кулаки ребенка сжаты.
Поздние проявления:
- Атрофия зрительных нервов вплоть до слепоты;
- Усиление сухожильных рефлексов;
- Мышечный спастический тетрапарез;
- Миоклонические судороги;
- Общая двигательная реакция.
Периферическая нейропатия встречается только у отдельных детей. Летальный исход у детей прослеживается в возрасте от семи месяцев до трех лет.
Волокнистая лейкодистрофия Александера
Патогенетический механизм развития болезни Александера – дефект гена, отвечающего за выработку протеина GFAP. Дефект провоцирует избыточное скопление белка внутри глиальной ткани головного мозга. Уникальная структура протеина позволяет диагностировать нозологию посредством обнаружения специальных волокон Розенталя.
Неонатальная форма приводит к летальному исходу через 1 год после начала.
Менее опасен инфантильный вид, при котором возникают пороки развития, гидроцефалия, атаксия, парезы, спастическое сокращение мускулатуры. В большинстве случаев смерть возникает через пару лет.
Ювенильная дистрофия Александера появляется у школьников в возрасте 4-10 лет. Стволовая симптоматика длится долго. Симптоматика прогрессирует на протяжении 10-20 лет. Манифестация во взрослом периоде имеет медленное течение. Общая продолжительность заболевания свыше 10 лет.
Лейкодистрофия Галлервордена-Шпатца
Начинается заболевания у детей в возрасте 10 лет.
Клинические симптомы патологии:
- Эпилептические приступы;
- Тетрапарез;
- Дисфункция стриопаллидарной сферы;
- Ретинит пигментный;
- Гимералопия.
Поздняя форма, возникающая у детей в школьные годы. Длительность нозологии до полного появления клинических проявлений – около десяти лет.
Основные признаки:
- Эпилептические припадки;
- Судорожные подергивания;
- Ригидность мускулатуры;
- Гиперкинетические состояния.
Передается патология по аутосомно-рецессивному типу. Возникает у лиц женского и мужского пола. Сопровождается выраженным слабоумием, полной обездвиженностью пациентов. Патоморфологические изменения:
- Избыточное накопление железа внутри тканей;
- Инфильтративные скопления в глиальном слое;
- Дегенеративные поражения аксонов;
- Повышенная пигментация таламуса, мозжечка, коры большого мозга, субталамических структур;
- Расстройство пигментно-липидного обмена;
- Дисбаланс катехоламинов.
Паталогоанатомическое обследование выявляет морфологические признаки.
Наследуется по аутосомно-рецессивному механизму.
Болезнь Нимана-Пика
Сфингомиелиновые расстройства типов A и B возникают по причине недостаточности фермента – сфингомиелиназы. Соединение необходимо для разрушения сфингомиелина.
Симптомы болезни Нимана-Пика:
- Расширение селезенки, поджелудочной железы, печени;
- Покраснение внутриглазной сетчатки;
- Неврологические расстройства;
- Ожирение внутренних органов.
Сфингомиелиновый жировой липидоз приводит к постепенному поражению паренхиматозных структур (почки, печень, селезенка).
Болезнь Гоше
Нозология характеризуется липидозом, сопровождающимся недостаточностью фермента глюкозилцерамидазы. Ранние стадии сопровождаются гепатоспленомегалией. Болевых ощущений, другой симптоматики не возрастает до тех пор, пока размеры органов не станут огромными.
Прогрессирующие неврологические расстройства обуславливают ранний летальный исход.
Разновидность патологии у взрослых людей обусловлена аутосомно-рецессивным механизмом передачи. Передача из поколения в поколение не доказана, но практика показывает вероятность информации.
Болезнь Гоше относится к категории взрослых заболеваний, но первые изменения появляются у детей в возрасте 10 лет. В более раннем или позднем возрасте симптоматика возникает значительно реже. Гиперспления, патологические переломы, асептические некрозы головки бедренной кости, псевдоостеомиелит – распространенные вторичные состояния на фоне первичной лейкодистрофии Гоше.
При всех разновидностях нозологии в костномозговом пунктате выявляются специальные «нагруженные клетки».
Болезнь Фабри
Патология встречается из-за дефекта фермента альфа-галактозидазы. В тканях избыточно скапливается вещество – тригексозид. Наследуется нозология по Х-хромосоме, поэтому часто встречается у мужчин.
Обычно формируется патология в пожилом возрасте. Клиническое проявление нозологии – болевая нейропатия. Магнитно-резонансная томография головного мозга не выявляет патологических изменений до возникновения прогрессирующего поражения почек. Средний возраст пациентов – 20-40 лет.
Артериальные тромбозы при болезни возникают в детском возрасте. Летальный исход формируется из-за выраженной недостаточности почек.
Болезнь Вольмана
Развивается у детей раннего возраста. Вначале прослеживается гепатоспленомегалия, затем присоединяются вторичные проявления:
- Рвотный рефлекс;
- Анемический синдром;
- Кальцинация надпочечников;
- Повышение концентрации холестерина;
- Фиброз печени.
Болезнь Вольмана передается по аутосомно-рецессивному типу.
Болезнь Краббе-Бенеке
Наследственная болезнь – лейкодистрофия Краббе передается аутосомно-рецессивным путем. Формируется нозология в детском возрасте, характеризуется рядом клинических признаков:
- Снижение слуха, зрения вплоть до полной слепоты;
- Деменция;
- Спастический паралич;
- Судороги мускулатуры;
- Децеребрационная ригидность.
Морфологические проявления нозологии сопровождаются демиелинизацией нервных оболочек, нарушением выработки церебролизидов. Лейкодистрофия Краббе генетически детерминирована. Клинические симптомы:
- Слепота;
- Снижение слуха;
- Мышечные спазмы;
- Судорожные припадки.
Носительство аномального гена обнаружить не удается. Отсутствует эффективное лечение.
Синонимы: диффузный инфантильный склероз, болезнь Краббе-Бенеке, глобоидно-клеточная лейкодистрофия.
Суданофильная лейкодистрофия Пелицеуса-Мерцбахера
Возникает нозология преимущественно у мальчиков, так как локализуется патологический ген в Х-хромосоме. Ученые не изучили патогенетические механизмы патологии. Диффузная демиелинизация обуславливает клинические проявления на первом году жизни. Возникает поражение стволовых структур головного и спинного мозга, мозжечка. Повреждение миелиновой оболочки приводит к разрушению центральных и периферических нервных волокон. На первом году жизни у человека возникают специфические признаки:
- Внутриглазной нистагм;
- Кивательное подергивание головы;
- Мышечные гипо- и гиперклонии;
- Паркинсонический синдром;
- Дегенерация волокон зрительного нерва;
- Снижение интеллектуальной функции.
Диффузная демиелинизация Пелицеуса-Мерцбахера наследуется по аутосомно-рецессивному механизму. Изменения серого вещества сопровождается повреждением осевых цилиндров.
Диагностика патологии на ранней стадии основана на первичных признаках:
- Нистагм;
- Нарушение координации;
- Дрожание головы.
Позднее присоединяется атрофия зрительного нерва, снижение интеллекта, мышечный гипертонус, нарушение речи. Тяжелая стадия патологии сопровождается нарастающей деменцией, паркинсоническим синдромом, гиперкинезами.
Перивентрикулярная лейкомаляция
Заболевание сопровождается повреждением белого вещества головного мозга. Характеризуется появлением некротических очагов с локализацией в перивентрикулярных сегментах. Сопровождается возникновением очагов некроза в полушариях, перивентрикулярных областях. Причина морфологических нарушений – гипоксически-ишемическая энцефалопатия. Клинические проявления нозологии:
- Задержка дыхания сразу после рождения;
- Снижение артериального давления;
- Повреждение белого вещества.
Возникновению нозологии у детей способствуют ишемические изменения. Возникает гипоксия, гипокапния, ацидоз у новорожденных детей из-за внутриутробной инфекции, длительных родов. Недостаток кислорода приводит к формированию очагов некроза с локализацией между вентрикулопетальными и вентриклофагальными артериальными ветвями.
Болезнь Канавана-ван-Богарта-Бертрана
Прогрессирующее повреждение нервных клеток головного мозга приводит к нейродегенеративным заболеваниям. Заболевание относится к ряду генетических изменений, приводящих к разрушению оболочки нейронов. Демиелинизация запускается геном, расположенным в семнадцатой хромосоме.
Комплекс морфологических изменений болезни Канавана провоцируется накоплением дефектного белка ASPA из-за недостатка фермента аспартоацилазы.
Симптомы лейкодистрофии:
- Умственная отсталость;
- Потеря моторной активности;
- Дефекты мышечного тонуса;
- Зрительная слепота;
- Трудности удержания головы в физиологической позиции.
Диагностика лейкодистрофии
Первоначальные признаки болезни выявляют клинические специалисты – педиатры, терапевты, неврологи, офтальмологи, отоларингологи.
Генетическое консультирование выявляет аномальные гены, провоцирующие сфинголипидозы головного мозга.
Клинические методы эхо-энцефалографии, нейросонографии выявляют увеличение внутричерепного давления. Исследование цереброспинальной жидкости проводится с целью обнаружения повышенной концентрации протеина.
Нарушение метаболизма выявляется биохимическими анализами крови.
МРТ головного мозга ребенку делают для определения очагов демиелинизации головного мозга. Исследование позволяет верифицировать патологические изменения ранней стадии.
Самый точный способ диагностики – инновационная ДНК-диагностика глобоидно-клеточной, метахроматической лейкодистрофии.